CB06
Z MBI
Obsah
Fylogenetické stromy
Terminológia:
- zakorenený strom, rooted tree
- nezakorenený strom, unrooted tree
- hrana, vetva, edge, branch
- vrchol, uzol, vertex, node
- list, leaf, leaf node, tip, terminal node
- vnútorný vrchol, internal node
- koreň, root
- podstrom, subtree, clade
Zopár faktov o stromoch
- Majme zakorenený strom s n listami, v ktorom má každý vnútorný vrchol 2 deti. Takýto strom vždy má n-1 vnútorných vrcholov a 2n-2 vetiev (prečo?)
- Majme nezakorenený strom s n listami, v ktorom má každý vnútorný vrchol 3 susedov. Takýto strom vždy má n-2 vnútorných vrcholov a 2n-3 vetiev
- Koľkými spôsobmi môžeme zakoreniť nezakorenený strom s n listami?
- koreň môže byť na hociktorej vetve stromu, teda je 2n-3 možností zakorenenia
- Ak nakreslíme zakorenený strom obvyklým spôsobom, listy sú usporiadané zhora nadol (alebo zľava doprava). Koľko rôznych poradí listov vieme dostať rôznym zakresľovaním toho istého stromu s n listami?
- máme n-1 vnutornych vrcholov, v kazdom mozeme vymenit lave a prave dieta. Pre kazdu konfiguraciu takychto vymen dostavame ine poradie, celkovy pocet poradi je 2n-1
- Čo vieme zistiť o príbuznosti organizmov z nezakoreneného stromu (napr. kvartet 4 organizmov)?
- skúsime zakoreniť všetkými spôsobmi a vidíme, že o žiadnych dvoch listoch nevieme povedať, že by boli sesterské (evolučne bližšie než ostatné), lebo koreň stromu môže byť zrovna na niektorej hrane vedúcej ku nim
- vieme však zistiť, že niektoré dvojice sesterské nebudú
Iné použitie stromov v informatike:
- uvidíme hierarchické zhlukovanie, bayesovské siete, ale tiež efektívne dátové štruktúry
Bootstrap
- Náhodne vyberieme niektoré stĺpce zarovnania, zostrojíme strom vybranou metódou
- Celé to opakujeme veľa krát
- Značíme si, koľkokrát sa ktorá vetva opakuje v stromoch, ktoré dostávame
- Pri nezakorenených stromoch je vetva rozdelenie listov na dve skupiny (bipartícia)
- Nakoniec zostavíme strom z celých dát a pozrieme sa ako často sa ktorá jeho vetva vyskytovala
- Môžeme zostaviť aj strom z často sa vyskytujúcich hrán (napr. tých, ktoré sú vo viac ako 50% stromov)
- Bootstrap hodnoty nám dajú určitý odhad spoľahlivosti, hlavne ak máme celkovo málo dát (krátke zarovnanie)
- Ak však dáta nezodpovedajú vybranej metóde/modelu, tak aj pre zlý strom môžeme dostať vysoký bootstrap
Detail: pri náhodnom výbere stĺpcov pre bootstrap väčšinou používame rovnaký počet stĺpcov ako pôvodné zarovnanie, pričom vzorkujeme z pôvodného zarovnania s povolením opakovania stĺpca.
Príklad

- Robili sme 100x bootstrap, 40x sme dostali strom (i) na obrázku, 40x sme dostali strom (ii) a 20x sme dostali strom (iii)
- Strom (iii) sme dostali aj spustením metody na celých dátach
- Zistite úroveň bootstrap podpory pre jednotlivé vetvy stromu (iii)
- Ktoré ďalšie vetvy majú podporu aspoň 20%?
- Aký strom by sme dostali, ak by sme chceli nechať iba vetvy s podporou aspoň 80%?
Opakovanie pravdepodobnostných modelov
Keď počítame pravdepodobnosť, rozmýšľame o myšlienkovom experimente, v ktorom hádžeme kockou, ťaháme gulôčky z vreca a pod.
- Dôležité je vždy si poriadne uvedomiť, ako tento experiment prebieha
- Tieto myšlienkové experimenty však nastavujeme tak, aby odzrkadľovali nejaké aspekty reality, napr. skutočných DNA sekvencií, ich evolúcie a pod.
- Takže pravdepodobnosti, ktoré spočítame v idealizovanom svete nám možno niečo povedia o reálnom svete
- Slávny citát štatistika Georga Boxa "All models are wrong, but some are useful."
Aké sme doteraz videli modely
- Skórovacie matice: porovnavame model nahodnych sekvencii a model nahodnych zarovnani
- E-value v BLASTe: nahodne vygenerujeme databazu a dotaz (query), kolko bude v priemere medzi nimi lokalnych zarovnani so skore aspon T?
- Hladanie genov: model generujuci sekvenciu+anotaciu naraz (parametre nastavene na znamych genoch). Pre danu sekvenciu, ktora anotacia je najpravdepodobnejsia?
- Evolucia, Jukes-Cantorov model: model generujuci stlpec zarovnania. Nezname parametre: strom, dlzky hran. Pre danu sadu stlpcov zarovnania, ktore parametre povedu k najvacsej pravdepodobnosti?
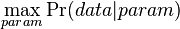
- Trochu detailov: pravdepodobnost zmeny/nezmeny na hrane dlzky t:
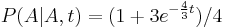 ,
, 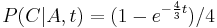
- Ak pozname ancestralne sekvencie, vieme spocitat pravdepodobnost dat
- Ancestralne sekvencie su nahodne premenne, ktore nas nezaujimaju: marginalizujeme ich (uvazujeme vsetky ich mozne hodnoty)
- Trochu detailov: pravdepodobnost zmeny/nezmeny na hrane dlzky t:
Zložitejšie evolučné modely
- Jukes-Cantorov model uvažuje len dĺžku hrany udanú ako priemerný počet substitúcií (vrátane tých, ktore nevidíme, kvôli tomu, že boli dve na tom istom mieste)
- Nie všetky substitúcie sa dejú rovnako často: tranzície (v rámci pyrimidínov T<->C, v rámci purínov A<->G) sú pravdepodobnejšie ako tranzverzie (A,G)<->(C,T)
- Nie všetky nukleotidy sa v danom genóme vyskytujú rovnako často (napr. mitochondriálne genómy majú nízky obsah GC)
- Tieto javy zachytáva HKY model (Hasegawa, Kishino & Yano)
- Matica rýchlostí zmeny (substitution rate matrix)
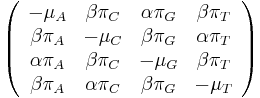
-
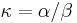 je pomer rychlosti, ktorymi sa deju tranzicie vs. transverzie
je pomer rychlosti, ktorymi sa deju tranzicie vs. transverzie
-
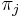 je frekvencia bazy
je frekvencia bazy  v sekvencii
v sekvencii
- Rychlost, ako sa deje substitucia z X do Y je sucin pravdepodobnosti Y a faktoru, ktory zavisi od toho, ci ide o tranziciu alebo transverziu
- Sucet kazdeho riadku matice ma byt 0, t.j.
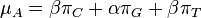
- Matica sa znormalizuje tak, aby priemerny pocet substitucii za jednotku casu bol 1
- Matica ma styri parametre:
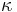 a tri frekvencie (stvrta musi doplnit do 1) plus dlzka hrany
a tri frekvencie (stvrta musi doplnit do 1) plus dlzka hrany
- Zlozitejsi model lepsie zodpoveda skutocnym procesom, ale na odhad viac parametrov potrebujeme viacej dat.
- Existuju metody, ktore pre dany cas t z matice rychlosti spocitaju pravdepodobnost, ze baza X zmutuje na bazu Y Pr(Y|X,t)
- Napr. pre velmi maly cas
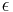 mame
mame 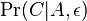 je zhruba
je zhruba 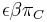
- Pre rozumne dlhe casy toto neplati, preto sa pouzivaju algebraicke metody, ktore beru do uvahy moznost viacerych substitucii na tom istom mieste
- Je aj vela inych modelov s mensim alebo vacsim poctom parametrov
Praktická ukážka tvorby stromov
- V UCSC browseri mozeme ziskavat viacnasobne zarovnania jednotlivych genov (nukleotidy alebo proteiny). Nasledujuci postup nemusite robit, subor si stiahnite tu: http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb06/cb06-aln.fa
- UCSC browseri si pozrieme usek ludskeho genomu (verzia hg38) chr6:135,851,998-136,191,840 s genom PDE7B (phosphodiesterase 7B) [1]
- Na modrej liste zvolime Tools, Table browser. V nastaveniach tabuliek Group: Genes and Gene Predictions, Track: GENCODE v 32., zaklikneme Region: position, a Output fomat: CDS FASTA alignment a stlacime Get output
- Na dalsej obrazovke zaklikneme show nucleotides, zvolime MAF table multiz100way a vyberieme si, ktore organizmy chceme. V nasom pripade z primatov zvolime chimp, rhesus, bushbaby, z inych cicavcov mouse, rat, rabbit, pig, cow, dog, elephant a z dalsich organizmov opposum, platypus, chicken, stlacime Get output.
- Vystup ulozime do suboru, nechame si iba prvu formu genu (ENST00000308191.11_hg38), z mien sekvencii zmazeme spolocny zaciatok (ENST00000308191.11_hg38), pripadne celkovo prepiseme mena na anglicke nazvy
- Skusme zostavit strom na stranke http://www.ebi.ac.uk/Tools/phylogeny/clustalw2_phylogeny/
- Distance correction: ako na prednáške, z počtu pozorovaných mutácií na evolučný čas
- Exclude gaps: vynechať všetky stĺpce s pomlčkami
- Clustering method: UPGMA predpokladá molekulárne hodiny, spájanie susedov nie
- P.I.M. vypíš aj maticu vzdialeností (% identity, pred korekciou)
- Vo vyslednom strome by sme mali zmenit zakorenenie, aby sme mali sliepku (chicken) ako outgroup
- Výsledky z programu http://www.phylogeny.fr/alacarte.cgi , ktorý podporuje aj bootstrap:
- Vysledok s povodnym zakorenenim
- Vysledok so spravnym zakorenenim (chicken = outgroup)
- "Spravny strom" [2] v nastaveniach Conservation track-u v UCSC browseri (podla clanku Murphy WJ et al Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001 [3])
- Nas strom ma dost zlych hran: zle postavenie hlodavcov, ale aj slona a psa. Zle postavenie hlodavcov môže byť spôsobené long branch attraction.
- Ak chcete skusit zostavit aj zarovnania, treba zacat z nezarovnanych sekvencii: [4]
![[2]](http://genome-euro.ucsc.edu/images/phylo/hg38_100way.png){kind=link}
Ďalšia zaujímavá webstránka s veľa nástrojmi https://usegalaxy.eu/
- na tvorbu stromov sa dá použiť IQ-TREE
- modely vid tu: https://github.com/Cibiv/IQ-TREE/wiki/Substitution-Models
- vysledok #CB:phylo
- viewer napr. http://phylotree.hyphy.org/
Fitchov algoritmus
Nerobili sme, uvedene pre zaujimavost

- Parsimony/uspornost
- Vstup: fylogeneticky strom, 1 stlpec zarovnania (jedna baza v kazdom liste stromu)
- Vystup: priradenie baz predkom minimalizujuce pocet substitucii
- Priklad - obr 1
- Uvazujme, co vieme povedat o strome s dvoma susednymi listami vo vacsom strome (oznacenie: obr. 2, listy v1 a v2, hrany do listov e1, e2, ich predok v3, hrana z v3 vyssie e3).
- Ak oba listy maju bazu rovnaku bazu, napr. A, predok v3 v optimalnom rieseni bude urcite mat bazu A
- Dokaz sporom: nech to tak nie je, nech optimalne riesenie ma nejaku inu bazu, napr. C. Vymenme v tomto rieseni toto C za A. Moze nam pribudnut jedna mutacia na hrane e3, ale ubudnu dve na hranach e1 a e2. Tym celkova cena riesenia klesne o 1, takze nebolo optimalne.
- Ak tieto dva listy maju rozne bazy, napr. A a C, tak existuje optimalne riesenie, ktore ma v predkovi v3 bazu A alebo C.
- Dokaz: vezmime optimalne riesenie. Ak ma v3 bazu A alebo C, tvrdenie plati. Ak ma v3 nejaku inu bazu, napr T, mozeme ju vymenit napr. za A, ci mozno pribudne jedna mutacia na e3 ale urcite ubudne mutacia na e1. Teda celkovy pocet mutacii sa nezvysi a nase nove riesenie je stale optimalne. Pozor, vo vseobecnosti nevieme povedat, ci ma v3 mat bazu A alebo C. V niektorych pripadoch su optimalne obe, v niektorych len jedna z nich.
- Fitchov algoritmus 1971
- Kazdemu vrcholu v priradime mnozinu baz M(v)
- M(v) pocitame od listov smerom ku korenu
- Pre list v bude M(v) obsahovat bazu v tomto liste
- Uvazujme vnutorny vrchol v s detmi x a y. Mame uz spocitane M(x) a M(y), chceme M(v)
- Ak M(x) a M(y) maju nejake spolocne bazy, vsetky tieto spolocne bazy dame do M(v), t.j.
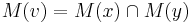
- Ak M(x) a M(y) nemaju spolocne bazy, do M(v) dame vsetky bazy z M(x) aj M(v), t.j.
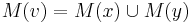
- V tomto pripade pocet mutacii vzrastie o jedna
- Ked mame M(v) spocitane pre vsetky vrcholy, ideme od korena smerom k listom a vyberieme vzdy jednu bazu z M(v).
- Ak sme vybrali pre rodica bazu x a x je v M(v), zvolime x aj pre v, inak zvolime lubovolnu bazu z M(v).
- Priklad algoritmu na obr 3