MBI 2010/2011
Z MBI
Obsah
UCSC genome browser, cvičenia pre biológov
- Používajte čierne počítače, zvoľte v menu Bioinformatika, užívatel Bioinformatika
- V programe Firefox choďte na stránku UCSC genome browser http://genome.ucsc.edu
- Hore v modrom menu zvoľte Genomes
- Na ďalšej stránke zvoľte človeka a v menu Assembly zistite, kedy boli pridané posledné dve verzie ľudského genómu (hg18 a hg19)
- Na tej istej stránke dole nájdete stručný popis zvolenej verzie genómu. Pre ktoré chromozómy máme viacero alternatívnych verzií?
- Zadajte región chr21:31,200,000-31,400,000
- Zapnite si track Mapability na "pack" a track RepeatMasker prepnite na "full"
- Mapability: nakoľko sa daný úsek opakuje v genóme a či teda vieme jednoznačne jeho ready namapovať pri použití Next generation sequencing (detaily keď kliknete na linku "Mapability")
- Približne v strede zobrazeného regiónu je pokles mapovateľnosti. Akému typu opakovania zodpovedá? (pozrite track RepeatMasker)
- Zapnite si tracky "Assembly" a "Gaps" a pozrite si región chr2:110,000,000-110,300,000. Aká dlhá je neosekvenovaná medzera (gap) v strede tohto regiónu? Približnú veľkosť môžete odčítať z obrázku, presnejší údaj zistíte kliknutím na čierny obdĺžnik zodpovedajúci tejto medzere (úplne presnú dĺžku aj tak nepoznáme, nakoľko nie je osekvenovaná).
- Prejdite na genóm Rhesus, región chr7:59,022,000-59,024,000, zapnite si tracky Contigs, Gaps, Quality scores
- Aké typy problémov v kvalite sekvencie v tomto regióne vidíte?
Sekvenovanie genómov, cvičenia pre informatikov
Uvod do pravdepodobnosti, pocitanie pokrytia genomov
- Nas problem: spocitanie pokrytia
- G = dlzka genomu, napr. 1 000 000
- N = pocet segmentov (readov), napr. 10 000
- L = dlzka readu, napr. 1000
- Celkova dlzka segmentov NL, pokrytie (coverage) NL/G, v nasom pripade 10x
- V priemere kazda baza pokryta 10x
- Niektore su ale pokryte viackrat, ine menej.
- Zaujimaju nas otazky typu: kolko baz ocakavame, ze bude pokrytych menej ako 3x?
- Dolezite pri planovani experimentov (aku velke pokrytie potrebujem na dosiahnutie urcitej kvality)
- Uvod do pravdepodobnosti
- Myslienkovy experiment, v ktorom vystupuje nahoda, napr. hod idealnou kockou/korunou
- Vysledkom experimentu je nejaka hodnota (napr. cislo, alebo aj niekolko cisel, retazec)
- Tuto neznamu hodnotu budeme volat nahodna premenna
- Zaujima nas pravdepodobnost, s akou nahodna premenna nadobuda jednotlive mozne hodnoty
- T.j. ak experiment opakujeme vela krat, ako casto uvidime nejaky vysledok
- Priklad 1: hodime idealizovanou kockou, premenna X bude hodnota, ktoru dostaneme
- Mozne hodnoty 1,2,..,6, kazda rovnako pravdepodobna
- Piseme napr. Pr(X=2)=1/6
- Priklad 2: hodime 2x kockou, nahodna premenna X bude sucet hodnot, ktore dostaneme
- Mozne hodnoty: 2,3,...,12
- Kazda dvojica hodnot na kocke rovnako pravdepodobna, t.j. pr. 1/36
- Sucet 5 mozeme dostat 1+4,2+3,3+2,4+1 - t.j. P(X=5) = 4/36
- Sucet 11 mozeme dostat 5+6 alebo 6+5, t.j. P(X=11) = 2/36
- Rozdelenie pravdepodobnosti: 2: 1/36, 3:2/36, 4: 3/36, ... 7: 6/36, 8: 5/36 ... 12: 1/36
- Sucet tychot hodnot je 1
- Pokrytie genomu: predpokladame, ze kazdy segment zacina na nahodnej pozicii zo vsetkych moznych G-L+1
- Takze ak premenna Y_i bude zaciatok i-teho segmentu, jej rozdelenie bude rovnomerne
- P(Y_i=1) = P(Y_i=2) = ... = P(Y_i=G-L+1) = 1/G-L+1
- Uvazujme premennu X_j, ktora udava pocet segmentov pokryvajucich poziciu j
- mozne hodnoty 0..N
- i-ty segment pretina poziciu j s pravdepodobnostou L/(G-L+1), oznacme tuto hodnotu p
- to iste ako keby sme N krat hodili mincou, na ktorej spadne hlava s pravd. p a znak 1-p a oznacili ako X_j pocet hlav
- taketo rozdelenie pravdepodobnosti sa vola binomicke
- P(X_j = k) = (N choose k) p^k (1-p)^(N-k)
- Zle sa pocita pre velke N, preto sa niekedy pouziva aproximacia Poissonovym rozdelenim s parametrom lambda = Np, ktore ma
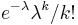
- Spat k sekvenovaniu: vieme spocitat rozdelenie pravdepodobnosti a tiez napr. P(X_i<3) = P(X_i=0)+P(X_i=1)+P(X_i=2) = 0.000045+0.00045+0.0023=0.0028 (v priemere ocakavame 45 baz nepokrytych, 2800 s menej ako 3 segmentami)
- Takyto graf, odhad, vieme lahko spravit pre rozne pocty segmentov a tak naplanovat, kolko segmentov potrebujeme
- Chceme tiez odhadnut pocet kontigov (podla clanku Lander a Waterman 1988)
- Ak niekolko baz vobec nie je pokrytych segmentami, prerusi sa kontig
- Vieme, kolko baz je v priemere nepokrytych, ale niektore mozu byt vedla seba
- Novy kontig vznikne aj ak sa susedne segmenty malo prekryvaju
- Predpokladajme, ze na spojenie dvoch segmentov potrebujeme prekryv aspon T=50
- Aka je pravdepodobnost, ze dany segment i bude posledny v kontigu?
- Ziaden segment j!=i nesmie zacinat v prvych L-T bazach kontigu i
- Kazdy segment tam zacina s pravdepodobnostou (L-T)/G, v priemere ich tam zacne N(L-T)/G
- Pouzijeme Poissonovo rozdelenie pre
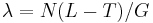 a k=0, t.j. pravdepodobnost, ze tam nezacne ziaden je zhruba exp(-N(L-T)/G)
a k=0, t.j. pravdepodobnost, ze tam nezacne ziaden je zhruba exp(-N(L-T)/G)
- Pre N segmentov dostaneme priemerny pocet kontigov N*exp(-N(L-T)/G)
- Ako keby sme dlzku segmentu skratili o dlzku prekryvu
- Pre T=50 dostaneme priemerny pocet kontigov 0.75 (v skutocnosti vzdy aspon 1, my sme vsak neuvazovali vplyv konca sekvencie). Ak znizime N na 5000 (5x pokrytie) dostaneme 43 kontigov
- Tento jednoduchy model nepokryva vsetky faktory:
- Segmenty nemaju rovnaku dlzku
- Problemy v zostavovani kvoli chybam, opkaovaniam a pod.
- Segmenty nie su rozlozene rovnomerne (cloning bias a pod.)
- Vplyv koncov chromozomov
- Uzitocny ako hruby odhad
- Na spresnenie mozeme skusat spravit zlozitejsie modely, alebo simulovat data
- Poznamka: pravdepodobnosti z binomickeho rozdelenia mozeme lahko spocitat napr. statistickym softverom R. Tu su prikazy, ktore sa na to hodia, pre pripad, ze by vas to zaujimalo:
dbinom(10,1e4,0.001); #(12.5% miest ma pokrytie presne 10) pbinom(10,1e4,0.001,lower.tail=TRUE); #(58% miest ma pokrytie najviac 10) dbinom(0:30,1e4,0.001); #tabulka pravdepodobnosti [1] 4.517335e-05 4.521856e-04 2.262965e-03 7.549258e-03 1.888637e-02 [6] 3.779542e-02 6.302390e-02 9.007019e-02 1.126216e-01 1.251601e-01 [11] 1.251726e-01 1.137933e-01 9.481826e-02 7.292252e-02 5.207187e-02 [16] 3.470068e-02 2.167707e-02 1.274356e-02 7.074795e-03 3.720595e-03 [21] 1.858621e-03 8.841718e-04 4.014538e-04 1.743354e-04 7.254524e-05 [26] 2.897743e-05 1.112843e-05 4.115040e-06 1.467156e-06 5.050044e-07 [31] 1.680146e-07
Zhrnutie
- Pravdedpobnostny model: myslienkovy experiment, v ktorom vystupuje nahoda, napr. hod idealizovanou kockou
- Vysledok je hodnota, ktoru budeme volat nahodna premenna
- Tabulka, ktora pre kazdu moznu hodnotu nahodnej premennej urci je pravdepodobnost sa vola rozdelenie pravdepodobnosti, sucet hodnot v tabulke je 1
- Znacenie typu P(X=7)=0.1
- Priklad: mame genom dlzky G=1mil., nahodne umiestnime N=10000 segmentov dlzky L=1000
- Nahodna premenna X_i je pocet segmentov pokryvajucich urcitu poziciu i
- Podobne, ako keby sme N krat hodili kocku, ktora ma cca 1 promile sancu padnu ako hlava a 99.9% ako znak a pytame sa, kolko krat padne znak (1 promile sme dostali po zaukruhleni z L/(G-L+1))
- Rozdelenie pravdepobnosti sa v tomto pripade vola binomicke a existuje vzorec, ako ho spocitat
- Takyto model nam moze pomoct urcit, kolko segmentov potrebujeme osekvenovat, aby napr. aspon 95% pozicii bolo pokrytych aspon 4 segmentami
Zostavovanie genomu pomocou eulerovskych tahov
- Opakovanie z teorie grafov:
- Hamiltonovska kruznica: cyklus, ktory prechadza kazdym vrcholom prave raz
- Eulerov tah: tah, ktory prechadza po kazdej hrane prave raz
- Zistit, ci ma graf H.c. je NP-tazke
- Problem obchodneho cestujuceho: najst najlacnejsiu H.c. je tiez NP-tazky
- Zistit ci ma graf E.t. a najst ho je lahke
- neorientovany graf na E.t. <=> je suvisly a vsetky vrcholy okrem najviac dvoch maju parny stupen
- orientovany graf ma E.t. z u do v <=> po pridani hrany z v do u je silne suvisly a vsetky vrcholy maju rovnako vchadzajucich ako vychadzajucich hran
- Najkratsie spolocne nadslovo: zjednodusena verzia problemu zostavovania genomov zo segmentov
- Mame danu mnozinu retazcov, chceme zostavit najkratsi retazec, ktoremu su vsetky podslova
- Priklad: GCCAAC,CCTGCC,ACCTTC zlozime v poradi 2,1,3: CCTGCCAACCTTC
- Mozeme si predstavit problem ako obmenu problemu obchodneho cestujuceho:
- retazcom priradim vrcholy
- dlzka hrany z a do b je
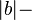 prekryv medzi a a b
prekryv medzi a a b
- specialny zaciatocny vrchol, z ktoreho hrany do kazdeho a s cenou |a|
- specialny koncovy vrchol, do ktoreho hrana z kazdeho a s cenou 0
- hrana z koncoveho do zaciatocneho vrcholu na uzavretie cyklu
- Hamiltonovske kruznice zodpovedaju nadslovam
- Ak najdeme najlacnejsiu Hamiltonovsku kruznicu, mame najkratsie nadslovo
- Ale vieme, ze problem obchodneho cestujuceho je NP-tazky
- Je toto dokaz, ze aj najkratsie spolocne nadslovo je NP-tazke?
- Pevzner, Tang and Waterman 2001 navrhuju namiesto Hamiltonovskej kruznice pouzit Eulerov tah (na inom grafe)
- deBruijnov graf stupna k:
- vrcholy: podretazce dlzky k vsetkych vstupnych retazcov
- hrany: nadvazujuce k-tice v ramci kazdeho segmentu (s prekryvom k-1)
- Priklad, k=2
- Chceme prejst po vsetkych hranach, chceme chodit co najmenej, takze po kazdej len raz - Eulerov tah
- V com je finta? Ako sme sa dostali od NP-tazkeho problemu k lahkemu?
- Co ak deBruijnov graf nema Eulerovsky tah? Znasobime niektore hrany, tieto budu zodpovedat opakovaniam
- Čo ak de Bruijnov graf má viacero Eulerovských ťahov?
- Zoberieme taký ťah, ktorý obsahuje pôvodné segmenty ako podcesty
- Zase ťažký problém, ale v praxi pomáhajú jednoduché pravidlá
- Opatrné riešenie: Ak z vrcholu 2 cesty, rozdeľ na kontigy
- Pouzitie sparovanych segmentov:
- Nájdi vrcholy v grafe, ktoré im zodpovedajú
- Ak je v grafe jediná cesta medzi týmito vrcholmi vhodnej dĺžky, premeň spárované segmenty na jeden veľký segment
- Dalsie problemy, ktore treba riesit
- sekvenovacie chyby: vytvaraju "bubliny" alebo slepe cesty
- dve vlakna
- Realne pouzivana technologia, aj pre sekvenovanie novej generacie, napr. Zerbino and Birney 2008 program Velvet
Počítanie fylogenetických stromov, cvičenia pre informatikov
- Ako definujeme strom v teorii grafov? suvisly acyklicky neorientovany graf
- Strom s n vrcholmi ma n-1 hran
- Nezakoreneny binarny fylogeneticky strom: neorientovany suvisl acyklicky graf, v listoch sucasne druhy, vsetky vnutorne vrcholy stupna 3
- Zakoreneny binarny fylogeneticky strom: vsetky hrany orientujeme od korena smerom k listom, kazdy vnutorny vrchol ma dve deti
- Niekedy uvazujeme aj nebinarne stromy, v ktorych mame vnutorne vrcholy vyssieho stupna
- Zakoreneny binarny strom s n listami ma n-1 vnutornch vrcholov, teda 2n-2 hran
- Nezakoreneny binarny strom s n listami ma n-2 vnutornych vrcholov, teda 2n-3 hran
- Pocet nezakorenenych fylogenetickych stromov s n listami:
- a(3) = 1, a(4) = 3, a(n+1) = a(n) * (2n-3) a teda a(n) = 1 * 3 * 5 * ... * (2n-5) = (2n-5)!!
- Pocet zakorenenych fylogenetickych stromov s n listami:
- zakoren strom s n listami kazdy 2n-3 sposobmi, teda (2n-3)!!