1-BIN-301, 2-AIN-501 Methods in Bioinformatics, 2023/24
CB08: Rozdiel medzi revíziami
Z MBI
(Vytvorená stránka „Na týchto cvičeniach sa budeme venovať trom štatistickým témam súvisiacim s komparatívnou genomikou a s analýzou expresie génov. Tieto techniky sa však využ...“) |
(→Multiple testing correction) |
||
| Riadok 117: | Riadok 117: | ||
* Preto potrebujeme pri velkom mnozstve testov umelo znizit prah na p-value tak, aby nahodny sum netvoril velke percento nasich vysledkov | * Preto potrebujeme pri velkom mnozstve testov umelo znizit prah na p-value tak, aby nahodny sum netvoril velke percento nasich vysledkov | ||
* Toto sa vola multiple testing correction, je viac technik, napr. FDR (false discovery rate) | * Toto sa vola multiple testing correction, je viac technik, napr. FDR (false discovery rate) | ||
| + | |||
| + | ==Nadreprezentácia, Uniprot (cvičenie pri počítači)== | ||
| + | Data o expresii ludskych genov v roznych tkanivach a podobne v '''UCSC genome browseri''' | ||
| + | * Chodte na genome browser http://genome-euro.ucsc.edu/ | ||
| + | * Zvolte ''Tools->Gene Sorter'', ''sort by'' nechajme ''Expression (GTEx)'', a do okienka ''search'' zadajme identifikator genu ''PTPRZ1'' | ||
| + | ** Dostane tabulku genov s podobnym profilom expresie ako PTPRZ1 (červená je vysoká expresia, zelená nízka) | ||
| + | ** Zoznam tychto genov v textovom formate najdete [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb08/zoznam_genov.txt tu] | ||
| + | * http://biit.cs.ut.ee/gprofiler/ mena genov skopirujme do policka ''Query'', stlacte g:Profile! | ||
| + | ** Ak by výpočet dlho trval, nájdete ho aj [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb08/g_Profiler.html tu] | ||
| + | ** Vo výslednej tabuľke je každý riadok jedna funkcna kategoria, v ktorej su geny s tymto profilom expresie nadreprezentovane, kazdy stlpec jeden gen. | ||
| + | ** V spodnej casti tabuly su aj asociacie k chorobam a k transkripcnym faktorom, ktore by mohli prislusne geny regulovat | ||
| + | * Co by sme na zaklade nadreprezentovanych kategorii usudzovali o gene PTPRZ1? | ||
| + | |||
| + | * Najdite tento gen v Uniprote (http://www.uniprot.org/), potvrdzuje nase domnienky? | ||
| + | ** O mnohých údajoch na stránke sme sa rozprávali na prednáške (GO kategórie, domény, sekundárna a 3D štruktúra) | ||
| + | ** na veľa miestach na stránke je uvedené aj odkiaľ jednotlivé údaje pochádzajú | ||
| + | ** Všimnime si Pfam domény a pozrime si ich stránku | ||
| + | |||
| + | * Vratme sa do genome browsera, najdime si PTPRZ1 gen v genome [http://genome-euro.ucsc.edu/cgi-bin/hgTracks?db=hg38&position=chr7%3A121873089-122062036] | ||
| + | * V browseri su rozne tracky tykajuce sa expresie, napr. GTEx. Precitajte si, co je v tomto tracku zobrazene, zapnite si ho a pozrite si expresiu okolitych genov okolo PTPRZ1 | ||
| + | * Kliknite na gen v tracku UCSC known genes. V tabulke uvidite zase prehlad expresie v roznych tkanivach (podla GTEx) | ||
Verzia zo dňa a času 10:21, 12. november 2020
Na týchto cvičeniach sa budeme venovať trom štatistickým témam súvisiacim s komparatívnou genomikou a s analýzou expresie génov. Tieto techniky sa však využívajú aj v iných oblastiach a môžete sa s nimi často stretnúť v genomických článkoch.
Obsah
Zhlukovanie
- Máme vstupné dáta, väčšinou ako vektory dĺžky n
- Snažíme sa ich rozdeliť do skupín tak, aby dáta v rámci skupiny boli podobné a medzi skupinami rôzne
Využitie:
- hľadanie génov s podobným profilom expresie
- hľadanie skupín pacientov s podobným profilom expresie génov (objavovanie podtypov nejakej choroby)
- hľadanie rodín podobných proteínov
- automatická segmentácia obrázkov (napríklad rozlíšiť jednotlivé políčka microarray alebo gelu od pozadia)
Na prednáške sme videli hierarchické zhlukovanie, ktoré z dát vytvorilo strom. Teraz si ukážeme zhlukovanie, ktoré sa snaží dáta rozdeliť na k skupín, kde k je vopred daný parameter.
K-Means


- pozri tiež prezentáciu pdf
- Vstup: n-rozmerné vektory
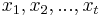 a počet zhlukov k
a počet zhlukov k
- Výstup: Rozdelenie vektorov do k zhlukov takéto:
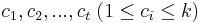 - priradenie vektoru k zhluku
- priradenie vektoru k zhluku
- n-rozmerné vektory
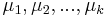 - centrá každého zhluku
- centrá každého zhluku
- Úloha: minimalizovať súčet štvorcov vzdialeností od každého vektoru k centru jeho zhluku:
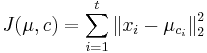
-
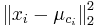 je druhá mocnina vzdialenosti vektora xi od centra jeho zhluku
je druhá mocnina vzdialenosti vektora xi od centra jeho zhluku
-
Algoritmus
Heuristika, ktorá nenájde vždy najlepšie zhlukovanie. Začne z nejakého zhlukovania a postupne ho zlepšuje. Pozri aj clanok na Wikipedii
- inicializácia: náhodne vyber k centier
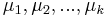
- opakuj kým sa niečo mení:
- priraď každý bod najbližšiemu centru:
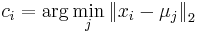
- vypočítaj nové centroidy:
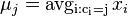 (spriemerujeme všetky body v jednom zhluku)
(spriemerujeme všetky body v jednom zhluku)
- priraď každý bod najbližšiemu centru:
Nadreprezentacia, obohatenie (enrichment)
- Mnohe celogenomove analyzy nam daju zoznam genov, ktore sa v nejakom ukazovateli vyrazne lisia od priemeru.
- Napriklad geny s pozitivnym vyberom v komparativnej genomike, geny vyrazne nadexprimovane alebo podexprimovane v microrarray experimentoch, geny regulovane urcitym transkripcnym faktorom a pod.
- Niektore z nich budu preskumanejsie (znama funkcia a pod.), niektore mozu mat nejake udaje o funkcii prenesene z homologov a dalsie mozu byt uplne nezname
- Co s takym zoznamom "zaujimavych genov"?
- moznost 1: vybrat si z neho niekolko malo zaujimavych kandidatov a preskumat ich podrobnejsie (experimentalne alebo informaticky)
- moznost 2: zistit, ci tato cela skupina je obohatena o geny urcitych skupin
- napr. v pripade pozitivneho vyberu nam casto vychadzaju geny suvisiace s imunitou, lebo su pod velkym evolucnym tlakom od patogenov
- takato analyza nam teda da informaciu o suvislostiach medzi roznymi procesmi
- Priklad (Kosiol et al)
- 16529 genov celkovo, 70 genov v GO kategorii innate immune response (0.4% zo vsetkych genov)
- 400 genov s pozivnym vyberom, mame 8 genov s innate immune response (2% zo vsetky genov s poz. vyb.)
- Celkovy pocet genov n, imunitnych ni, pozitivny vyber np, imunitnych s poz. vyb. nip.
- Kontingencna tabulka
| Pozitivny vyber | Bez poz. vyberu | Sucet | |
|---|---|---|---|
| Imunitne | 8 (nip) | 62 | 70 (ni) |
| Ostatne | 392 | 16067 | 16459 |
| Sucet | 400 (np) | 16129 | 16529 (n) |
- Nulova hypoteza: geny v nasom zozname boli nahodne vybrane z celeho genomu, t.j. ak v celom genome je frekvencia imunitnych genov ni/n (cca 0.4%), vo vzorke velkosti np (geny s pozitivnym vyberom) ocakavame cca np * (ni / n) imunitnych genov.
- aj v nulovej hypoteze vsak vzorka velkosti ni cisto nahodou moze obsahovat viac alebo menej takych genov.
- presnejsie mame urnu so ni (70) bielymi a n-ni (16459) ciernymi gulickami, vytiahneme nahodne np (400) guliciek, kolko bude medzi nimi bielych, nazvime tuto nahodnu premennu Xip
- v nasom priklade by sme ocakavali 1.7 genu s innate immune response, ale mame 8 (4.7xviac)
- Rozdelenie pravdepodobnosti Xip je hypergeometricke, t.j.
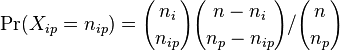
- Aka je pravdepodobnost, ze v nulovej hypoteze bude Xip tolko, kolko sme namerali alebo viac? (Chvost rozdelenia). V nasom pripade p-value 2.8e-4.
- Hypergeometric or Fisher's exact test, pripadne ich aproximacie pre velke hodnoty v tabulke (chi^2 test) zisti, ci sa nasa tabulka velmi lisi od toho, co by sme ocakavali v nulovej hypoteze
- Suvisiace clanky
- Rivals I, Personnaz L, Taing L, Potier MC (February 2007). "Enrichment or depletion of a GO category within a class of genes: which test?". Bioinformatics (Oxford, England) 23 (4): 401–7. doi:10.1093/bioinformatics/btl633. PMID 17182697.
- Huang da W, Sherman BT, Lempicki RA (January 2009). "Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists". Nucleic Acids Research 37 (1): 1–13. doi:10.1093/nar/gkn923. PMID 19033363.
- Reimand, Jüri, et al. "Pathway enrichment analysis and visualization of omics data using g: Profiler, GSEA, Cytoscape and EnrichmentMap." Nature protocols 14.2 (2019): 482. [1]
- Existuju web servery, napr. GOrilla pre ludske geny: http://cbl-gorilla.cs.technion.ac.il/, DAVID (http://david.niaid.nih.gov), g:Profiler http://biit.cs.ut.ee/gprofiler/
- Treba dat pozor, ci pocitaju to co chceme
- Kod v statistickom systeme R na pocitanie hypergeometrickeho rozdelenia
> dhyper(0:70, 70, 16529-70, 400); [1] 1.793421e-01 3.126761e-01 2.679872e-01 1.505169e-01 6.231088e-02 [6] 2.027586e-02 5.400796e-03 1.210955e-03 2.332580e-04 3.920215e-05 [11] 5.818723e-06 7.702558e-07 9.166688e-08 9.873221e-09 9.678760e-10 [16] 8.677204e-11 7.143849e-12 5.420388e-13 3.802134e-14 2.472342e-15 [21] 1.493876e-16 8.405488e-18 4.412274e-19 2.164351e-20 9.935473e-22 [26] 4.273662e-23 1.724446e-24 6.533742e-26 2.326517e-27 7.791092e-29 [31] 2.455307e-30 7.285339e-32 2.036140e-33 5.361856e-35 1.330660e-36 [36] 3.112566e-38 6.862558e-40 1.426089e-41 2.792792e-43 5.153006e-45 [41] 8.955105e-47 1.465159e-48 2.255667e-50 3.265636e-52 4.442631e-54 [46] 5.674366e-56 6.797781e-58 7.629501e-60 8.012033e-62 7.860866e-64 [51] 7.193798e-66 6.129013e-68 4.851139e-70 3.558526e-72 2.412561e-74 [56] 1.506983e-76 8.641725e-79 4.530590e-81 2.161126e-83 9.326620e-86 [61] 3.617279e-88 1.250737e-90 3.817900e-93 1.016417e-95 2.323667e-98 [66] 4.469699e-101 7.034762e-104 8.698702e-107 7.924236e-110 4.728201e-113 [71] 1.386176e-116 phyper(7, 70, 16529-70, 400, lower.tail=FALSE); # pr pocet bielych>7 (t.j. >=8) ak taham 400 z vreca so 70 bielymi a 16529-70 ciernymi # sucet cisiel z tabulky od 2.332580e-04 az po koniec d = dhyper(0:15, 70, 16529-70, 400); plot(0:15,d) # test pre danu tabulku a=matrix(c(8,62,392,16067),nrow=2, ncol=2) fisher.test(a,alternative = "greater")
Multiple testing correction
- V mnohych situaciach robime vela testov toho isteho typu, kazdy ma urcitu p-value
- Napr. testujeme 1000 genov v genome na pozitivny vyber, zvolime tie, kde p-value <= 0.05
- Alebo testujeme obohatenie 1000 funkcnych kategorii v nejakej vzorke genov, zvolime tie, kde p-value <= 0.05
- Problem: ak kazda z 1000 kategorii ma 5% sancu tam byt len nahodou, ocakavali by sme 50 cisto nahodnych pozitivnych vysledkov. Ak sme napr. nasli 100 pozitivnych vysledkov (obohatenych kategorii), cca polovica z nich je zle
- Preto potrebujeme pri velkom mnozstve testov umelo znizit prah na p-value tak, aby nahodny sum netvoril velke percento nasich vysledkov
- Toto sa vola multiple testing correction, je viac technik, napr. FDR (false discovery rate)
Nadreprezentácia, Uniprot (cvičenie pri počítači)
Data o expresii ludskych genov v roznych tkanivach a podobne v UCSC genome browseri
- Chodte na genome browser http://genome-euro.ucsc.edu/
- Zvolte Tools->Gene Sorter, sort by nechajme Expression (GTEx), a do okienka search zadajme identifikator genu PTPRZ1
- Dostane tabulku genov s podobnym profilom expresie ako PTPRZ1 (červená je vysoká expresia, zelená nízka)
- Zoznam tychto genov v textovom formate najdete tu
- http://biit.cs.ut.ee/gprofiler/ mena genov skopirujme do policka Query, stlacte g:Profile!
- Ak by výpočet dlho trval, nájdete ho aj tu
- Vo výslednej tabuľke je každý riadok jedna funkcna kategoria, v ktorej su geny s tymto profilom expresie nadreprezentovane, kazdy stlpec jeden gen.
- V spodnej casti tabuly su aj asociacie k chorobam a k transkripcnym faktorom, ktore by mohli prislusne geny regulovat
- Co by sme na zaklade nadreprezentovanych kategorii usudzovali o gene PTPRZ1?
- Najdite tento gen v Uniprote (http://www.uniprot.org/), potvrdzuje nase domnienky?
- O mnohých údajoch na stránke sme sa rozprávali na prednáške (GO kategórie, domény, sekundárna a 3D štruktúra)
- na veľa miestach na stránke je uvedené aj odkiaľ jednotlivé údaje pochádzajú
- Všimnime si Pfam domény a pozrime si ich stránku
- Vratme sa do genome browsera, najdime si PTPRZ1 gen v genome [2]
- V browseri su rozne tracky tykajuce sa expresie, napr. GTEx. Precitajte si, co je v tomto tracku zobrazene, zapnite si ho a pozrite si expresiu okolitych genov okolo PTPRZ1
- Kliknite na gen v tracku UCSC known genes. V tabulke uvidite zase prehlad expresie v roznych tkanivach (podla GTEx)