CI12: Rozdiel medzi revíziami
(→Protein threading) |
|||
| Riadok 34: | Riadok 34: | ||
* Chceme minimalizovat <math>\sum_{i=1}^n x_i\,</math> | * Chceme minimalizovat <math>\sum_{i=1}^n x_i\,</math> | ||
* za podmienky, ze pre kazde j z {1..m} plati <math>\sum_{i:j\in S_i} x_j \ge 1</math> | * za podmienky, ze pre kazde j z {1..m} plati <math>\sum_{i:j\in S_i} x_j \ge 1</math> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| Riadok 70: | Riadok 61: | ||
Zdroj: | Zdroj: | ||
* Jinbo Xu, Ming Li, Dongsup Kim, and Ying Xu. "RAPTOR: optimal protein threading by linear programming." Journal of bioinformatics and computational biology 1, no. 01 (2003): 95-117. [http://ttic.uchicago.edu/~jinbo/SelectedPubs/RAPTOR.pdf] | * Jinbo Xu, Ming Li, Dongsup Kim, and Ying Xu. "RAPTOR: optimal protein threading by linear programming." Journal of bioinformatics and computational biology 1, no. 01 (2003): 95-117. [http://ttic.uchicago.edu/~jinbo/SelectedPubs/RAPTOR.pdf] | ||
| + | |||
| + | ===Iný príklad: Zarovnanie sekvencií RNA so štruktúrou=== | ||
| + | |||
| + | Máme dané dve sekvencie RNA a pre každú z nich máme daný zoznam párov báz (pozícií v rámci sekvencie), ktoré by mohli byť prítomné v sekundárnej štruktúre. | ||
| + | * Zoznam párov môže byť konkrétna známa sekundárna štruktúra danej sekvencie alebo väčšia množina párov, ktoré by sa v štruktúre mohli vyskytovať, napríklad dvojice, ktoré majú pomerne veľkú pravdepodobnosť byť spárené v SCFG modeli alebo dokonca všetky dvojice komplementárnych báz. | ||
| + | |||
| + | Cieľom je nájsť optimálne zarovnanie týchto dvoch sekvencií, v ktorom použijeme obvyklé skórovanie zhôd, nezhôd a medzier, ale navyše pridáme nejaké skóre za zhody v štruktúre. Dva potenciálne páry, každý z jednej sekvencie, považujeme za zarovnané, ak sú navzájom zarovnané bázy na ich obidvoch koncoch. Do skórovania vyberieme podmnožinu zarovnaných párov tak, aby každá báza bola v najviac jednom páre a každému takému páru priradíme nejaké kladné skóre. | ||
| + | |||
| + | Detaily: | ||
| + | https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-8-271 | ||
Aktuálna revízia z 09:03, 14. december 2023
Obsah
Protein threading
Praktické programy na NP ťažké problémy
- Obcas chceme najst optimalne riesenie nejakeho NP-tazkeho problemu
- Jedna moznost je previest na iny NP tazky problem, pre ktory existuju pomerne dobre prakticke programy, napriklad integer linear programming (ILP)
- najdu optimalne riesenie, mnohe instancie zrataju v rozumnom case, ale mozu bezat aj velmi dlho
- CPLEX [1] a Gurobi [2] komercne baliky na ILP, akademicka licencia zadarmo
- SCIP [3] nekomercny program pre ILP
- SYMPHONY v projekte COIN-OR [4]
- Minisat [5] open source SAT solver, tiež Lingeling, glucose, CryptoMiniSat, painless
- Concorde TSP solver [6] - riesi problem obchodneho cestujuceho so symetrickymi vzdialenostami, zadarmo na akademicke ucely
- Pre zaujimavost: TSP art [7]
ILP
Lineárny program:
- Mame reálne premenné x_1...x_n, minimalizujeme nejaku ich linearnu kombinaciu
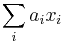 kde a_i su dane vahy.
kde a_i su dane vahy.
- Mame tiez niekolko podmienok v tvare linearnych rovnosti alebo nerovnosti, napr.
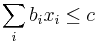
- Hladame teda hodnoty premennych, ktore minimalizuju cielovu sumu, ale pre ktore platia vsetky podmienky
- Da sa riesit v polynomialnom case
Integer linear program
- Program, v ktorom vsetky/vybrane premenne musia mat celociselne hodnoty, alebo dokonca povolime iba hodnoty 0 a 1.
- NP uplny problem
Ako zapisat (NP-tazke) problemy ako ILP
Knapsack
- Problem: mame dane predmety s hmotnostami w_1..w_n a cenami c_1..c_n, ktore z nich vybrat, aby celkova hmotnost bola najviac T a cena bola co najvyssia?
- Pouzijeme binarne premenne x_1..x_n, kde x_i = 1 prave vtedy ked sme zobrali i-ty predmet.
- Chceme maximalizovat
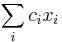
- za podmienky ze
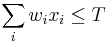
Set cover:
- Mame n mnozin S_1...S_n nad mnozinou {1...m}. Chceme vybrat co najmensi pocet zo vstupnych mnozin tak, aby ich zjednotenie bola cela mnozina {1..m}
- Binarne premenne x_i=1 ak vyberieme i-tu mnozinu
- Chceme minimalizovat
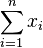
- za podmienky, ze pre kazde j z {1..m} plati
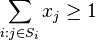
Protein threading
- Ciel: protein A ma znamu sekvenciu aj strukturu, protein B iba sekvenciu. Chceme zarovnat proteiny A a B, pricom budeme brat do uvahy znamu strukturu, t.j. ak su dve amino kyseliny blizko v A tak ich ekvivalenty v B by mali byt "kompatibilne".
- Tento problem chceme riesit tak, ze v strukture A urcime nejake jadra, ktore by v evolucii mali zostat zachovane bez inzercii a delecii a v rovnakom poradi. Tieto jadra su oddelene sluckami, ktorych dlzka sa moze lubovolne menit a ktorych zarovnania nebudeme skorovat.
- Formulacia problemu: Mame danu sekvenciu B=b1..bn, dlzky m jadier c_1...c_m a skorovacie tabulky E_ij, ktora vyjadruje, ako dobre bj..b_{j+c_i-1} sedi do sekvencie jadra i a E_ijkl ktora vyjadruje, ako dobre by jadra i a k interagovali, keby mali sekvencie zacinajuce v B na poziciach j a l. Uloha je zvolit polohy jadier x_1<x_2<...<x_m tak, aby sa ziadne dve jadra neprekryvali a aby sme dosiahli najvyssie skore.
- Poznamka: nevraveli sme, ako konkretne zvolit jadra a skorovacie tabulky, co je modelovaci, nie algoritmicky problem (mozeme skusit napr. nejake pravdepodobnostne modely)
Protein threading ako ILP
- Premenne v programe:
- x_ij=1 ak je zaciatok i-teho jadra zarovnane s b_j
- y_ijkl=1 ak je zaciatok i-teho jadra na b_j a zaciatok k-teho na b_l (i<k, j<l)
- Chceme maximalizovat
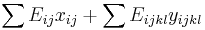
- Podmienky:
-
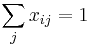 pre kazde i
pre kazde i
-
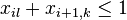 pre vsetky i,k,l, kde k<l+c_i
pre vsetky i,k,l, kde k<l+c_i
-
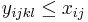 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
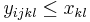 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
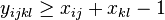 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
Na zamyslenie:
- Aka bude velkost programu ako funkcia n a m?
- Co ak nie vsetky jadra navzajom interaguju? Mozeme na velkosti programu usetrit?
- Preco asi vobec autori zaviedli jadra a ako by sme zmenili program, ak by sme chceli uvazovat kazdu aminokyselinu zvlast?
Zdroj:
- Jinbo Xu, Ming Li, Dongsup Kim, and Ying Xu. "RAPTOR: optimal protein threading by linear programming." Journal of bioinformatics and computational biology 1, no. 01 (2003): 95-117. [8]
Iný príklad: Zarovnanie sekvencií RNA so štruktúrou
Máme dané dve sekvencie RNA a pre každú z nich máme daný zoznam párov báz (pozícií v rámci sekvencie), ktoré by mohli byť prítomné v sekundárnej štruktúre.
- Zoznam párov môže byť konkrétna známa sekundárna štruktúra danej sekvencie alebo väčšia množina párov, ktoré by sa v štruktúre mohli vyskytovať, napríklad dvojice, ktoré majú pomerne veľkú pravdepodobnosť byť spárené v SCFG modeli alebo dokonca všetky dvojice komplementárnych báz.
Cieľom je nájsť optimálne zarovnanie týchto dvoch sekvencií, v ktorom použijeme obvyklé skórovanie zhôd, nezhôd a medzier, ale navyše pridáme nejaké skóre za zhody v štruktúre. Dva potenciálne páry, každý z jednej sekvencie, považujeme za zarovnané, ak sú navzájom zarovnané bázy na ich obidvoch koncoch. Do skórovania vyberieme podmnožinu zarovnaných párov tak, aby každá báza bola v najviac jednom páre a každému takému páru priradíme nejaké kladné skóre.
Detaily: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-8-271