CI05: Rozdiel medzi revíziami
(→Návrat do porovnávania sekvencií) |
|||
| Riadok 1: | Riadok 1: | ||
| − | == | + | ==Vzorec na vypocet senzitivity jadra== |
| + | * Uvazujme jadro dlzky w (ako v programe BLAST pre nukleotidy) | ||
| + | * Uvazujme pravdepodobnostny model zarovnania, v ktorom ma kazda pozicia pravdepodobnost p, ze bude zhoda a (1-p), ze bude nezhoda alebo medzera, zarovnanie ma dlzku L | ||
| + | * Nahodna premenna X_i = 1 ak na pozicii i je zhoda, 0 inak | ||
| + | * Nahodna premenna Y_i = 1 ak na pozicii i zacina jadro, t.j. ak <math>X_i=1, X_{i+1}=1, \dots, x_{i+w-1}=1</math> | ||
| + | * <math>P(Y_i = 1) = p^w</math>, nakolko X_i su navzajom nezavisle | ||
| + | * Nech <math>Y = \sum_{i=0}^{L-w} y_i</math> | ||
| + | * Z linearity strednej hodnoty vieme lahko odhadnut E(Y) = (L-w+1)p^w | ||
| + | * Nas ale zaujima P(Y>0) = 1-P(Y=0) | ||
| + | * <math>P(Y=0) = P(Y_0=0 \wedge \dots \wedge Y_{L-w}=0)</math> | ||
| + | * Preco neplati, <math>P(Y=0) = P(Y_i = 0)^{L-w+1}</math>? | ||
| + | ** Jendotlive Y_i nie su nezavisle, napr. <math>P(Y_{i+1}=1|Y_i=1)=p</math> | ||
| + | ** V postupnosti Y_i sa jendotky maju tendenciu klastrovat spolu | ||
| + | * P(Y>0) ale vieme spoctat dynamickym programovanim | ||
| + | * Nech A[L] je pravdepodobnost vyskytu jadra v zarovnani dlzky L | ||
| + | * <math> | ||
| + | A[L] = \left\{\begin{array}{ll} | ||
| + | 0 & \mbox{ak } L < w\\ | ||
| + | p^w+\sum_{i=0}^{w-1} p^i (1-p)A[L-i-1] & \mbox{ak } L \ge w\\ | ||
| + | \end{array}\right.</math> | ||
| − | + | ==Minimizery: ako usetrit pamat== | |
| + | * k-merom nazveme k za sebou iducich pismen (nukleotidov DNA) | ||
| + | * Zakladne pouzite jadier: pri porovnavani dvoch sekvencii (alebo mnozin sekvencii) uloz vsetky k-mery jednej sekvencie do slovnika (napr. hash tabulky), potom prechadzaj vsetky k-mery druhej sekvencie a hladaj ich v slovniku | ||
| − | == Odbočka do analýzy web-stránok: Podobnosť textov == | + | * Trik na znizenie potrebnej pamate (napr. program BLAT): ukladaj iba kazdy s-ty k-mer z prvej sekvencie, potom hladaj vsetky k-mery z druhej |
| + | * Trochu znizi aj senzitivitu, ale kedze jadra sa klastruju, mame sancu aspon jedno jadro z klastra najst | ||
| + | |||
| + | * Prefikanejsia idea je minimizer: uvazuj vsetky skupiny s po sebe iducich k-merov (sliding window), v kazdej skupine najdi abecedne najmensi k-mer (minimizer) a uloz do slovnika | ||
| + | * Pri posune okna o jedno doprava casto najmensi k-mer zostava ten isty a netreba ho znovu ukladat, cim sa usetri pamat | ||
| + | * Priklad k=5, s=4 | ||
| + | <pre> | ||
| + | AGTGGCTGCCAGGCTGG cGaGGCTGCCaGGtTGG | ||
| + | AGTGG* CGAGG | ||
| + | GTGGC GAGGC | ||
| + | TGGCT AGGCT* | ||
| + | GGCTG GGCTG | ||
| + | GCTGC* GCTGC | ||
| + | CTGCC* CTGCC* | ||
| + | TGCCA TGCCT | ||
| + | GCCAG GCCTG | ||
| + | CCAGG* CCTGG* | ||
| + | CAGGC* CTGGT* | ||
| + | AGGCT* TGGTT | ||
| + | GGCTG GGTTG | ||
| + | GCTGG GTTGG | ||
| + | </pre> | ||
| + | |||
| + | * Rozdiel je pri hladani: v slovniku nehladame vsetky k-mery druhej sekvencie, ale tiez len minimizery, co moze usetrit cas | ||
| + | * Obzvlast vyhodne ak prva a druha mnozina sekvencii su ta ista, napr. pri hladani prekryvov v citaniach pri skladani genomu. Kazde citanie ma mnozinu minimizerov, ktore sa pouziju ako kluce v slovniku, hodnoty su zoznamy citani. Dvojice citani zdilajuce nejaky minimizer (binning) sa dostanu do jedneho zoznamu a budu uvazovane pri vypocte vzajomneho prekryvu | ||
| + | * V praxi sa do slovnika neuklada lexikograficky najmensi k-mer, ale kazdy k-mer sa prehasuje nejakou funkciou f a zoberie sa ten s minimalnou hodnotou | ||
| + | * Dovod je, ze sa chceme vyhnut, aby minimazermi boli casto sekvencie typu AAAAA, ktore su v biologickych sekvenciach nadreprezentovane a casto nie su funkcne zaujimave | ||
| + | * Minimizery vyuziva napr. aj minimap2, velmi popularny nastroj na zarovnavanie citani navzajom a ku genomom | ||
| + | |||
| + | * Li, Heng. "Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences." Bioinformatics 32.14 (2016): 2103-2110. [https://academic.oup.com/bioinformatics/article-abstract/32/14/2103/1742895] | ||
| + | |||
| + | * Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. Reducing storage requirements for biological sequence comparison. Bioinformatics. 2004 Dec 12;20(18):3363-9. [https://academic.oup.com/bioinformatics/article/20/18/3363/202143] | ||
| + | |||
| + | |||
| + | ==MinHash== | ||
| + | === Odbočka do analýzy web-stránok: Podobnosť textov === | ||
Majme množinu webových stránok (webová stránka je postupnosť slov). Chceme nájsť medzi nimi dvojice podobných. Ako môžeme definovať podobnosť dvoch textov? | Majme množinu webových stránok (webová stránka je postupnosť slov). Chceme nájsť medzi nimi dvojice podobných. Ako môžeme definovať podobnosť dvoch textov? | ||
| Riadok 14: | Riadok 70: | ||
:::<math>J(A, B) := \dfrac{|A \cap B|}{|A \cup B|}</math> | :::<math>J(A, B) := \dfrac{|A \cap B|}{|A \cup B|}</math> | ||
| − | Táto miera nadobúda hodnotu 0 iba prípade, ak množiny sú disjunktné, a hodnotu 1 iba v prípade, že množiny sú totožné. Inak sa jej hodnota nachádza na otvorenom intervale <math>(0, 1)</math>, a čím viac spoločných slov majú, tým je jej hodnota vyššia. | + | Táto miera nadobúda hodnotu 0 iba v prípade, ak množiny sú disjunktné, a hodnotu 1 iba v prípade, že množiny sú totožné. Inak sa jej hodnota nachádza na otvorenom intervale <math>(0, 1)</math>, a čím viac spoločných slov majú, tým je jej hodnota vyššia. |
Potom otázku "Ktoré dvojice textov sú podobné?" môžeme preformulovať napríklad ako "Ktoré dvojice textov majú Jaccardovu mieru podobnosti vyššiu ako <math>\alpha</math>?", kde <math>\alpha \in (0, 1)</math> je nejaká prahová hodnota. | Potom otázku "Ktoré dvojice textov sú podobné?" môžeme preformulovať napríklad ako "Ktoré dvojice textov majú Jaccardovu mieru podobnosti vyššiu ako <math>\alpha</math>?", kde <math>\alpha \in (0, 1)</math> je nejaká prahová hodnota. | ||
| Riadok 20: | Riadok 76: | ||
Exaktný výpočet Jaccardovej miery podobnosti nie je vždy dostatočne rýchly pre účely konkrétnej aplikácie, takže logickým riešením je pokúsiť sa jej hodnotu vypočítať iba približne (t.j. aproximovať). | Exaktný výpočet Jaccardovej miery podobnosti nie je vždy dostatočne rýchly pre účely konkrétnej aplikácie, takže logickým riešením je pokúsiť sa jej hodnotu vypočítať iba približne (t.j. aproximovať). | ||
| − | == Aproximácia Jaccardovej miery: MinHash == | + | === Aproximácia Jaccardovej miery: MinHash === |
Nech je daná množina <math>A = \{a_1, a_2, \ldots, a_n\} \subseteq U</math>. Nech <math>h:U \to \mathbb{R}</math> je injektívna náhodná hashovacia funkcia (t.j. bez kolízií). Potom minimálny hash množiny <math>minHash_h(A)</math> je definovaný nasledovne: | Nech je daná množina <math>A = \{a_1, a_2, \ldots, a_n\} \subseteq U</math>. Nech <math>h:U \to \mathbb{R}</math> je injektívna náhodná hashovacia funkcia (t.j. bez kolízií). Potom minimálny hash množiny <math>minHash_h(A)</math> je definovaný nasledovne: | ||
| Riadok 58: | Riadok 114: | ||
Druhá možnosť zlepšenia je nechať jednu hashovaciu funkciu, ale porovnávať nie 1, ale k najmenších hashov dvoch množín. Vedie to ku podobnému asymptotickému správaniu. | Druhá možnosť zlepšenia je nechať jednu hashovaciu funkciu, ale porovnávať nie 1, ale k najmenších hashov dvoch množín. Vedie to ku podobnému asymptotickému správaniu. | ||
| − | == Návrat do porovnávania sekvencií == | + | === Návrat do porovnávania sekvencií === |
Ako "slová" použijeme všetky súvislé podreťazce fixnej dĺžky (dĺžka sa tradične označuje ako <math>k</math>) danej sekvencie. Tieto podreťazce sa tradične označujú ako <math>k</math>-mery. | Ako "slová" použijeme všetky súvislé podreťazce fixnej dĺžky (dĺžka sa tradične označuje ako <math>k</math>) danej sekvencie. Tieto podreťazce sa tradične označujú ako <math>k</math>-mery. | ||
Potom na hľadanie dvoch podobných čítaní z množiny čítaní môžeme použiť minhash. | Potom na hľadanie dvoch podobných čítaní z množiny čítaní môžeme použiť minhash. | ||
| − | |||
| − | |||
| − | |||
* Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome biology. 2016 Dec;17(1):1-4. [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0997-x] | * Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome biology. 2016 Dec;17(1):1-4. [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0997-x] | ||
| − | |||
| − | |||
| − | |||
| − | |||
Verzia zo dňa a času 21:36, 18. október 2021
Obsah
Vzorec na vypocet senzitivity jadra
- Uvazujme jadro dlzky w (ako v programe BLAST pre nukleotidy)
- Uvazujme pravdepodobnostny model zarovnania, v ktorom ma kazda pozicia pravdepodobnost p, ze bude zhoda a (1-p), ze bude nezhoda alebo medzera, zarovnanie ma dlzku L
- Nahodna premenna X_i = 1 ak na pozicii i je zhoda, 0 inak
- Nahodna premenna Y_i = 1 ak na pozicii i zacina jadro, t.j. ak

-
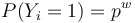 , nakolko X_i su navzajom nezavisle
, nakolko X_i su navzajom nezavisle
- Nech
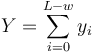
- Z linearity strednej hodnoty vieme lahko odhadnut E(Y) = (L-w+1)p^w
- Nas ale zaujima P(Y>0) = 1-P(Y=0)
-
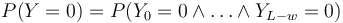
- Preco neplati,
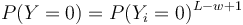 ?
?
- Jendotlive Y_i nie su nezavisle, napr.
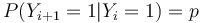
- V postupnosti Y_i sa jendotky maju tendenciu klastrovat spolu
- Jendotlive Y_i nie su nezavisle, napr.
- P(Y>0) ale vieme spoctat dynamickym programovanim
- Nech A[L] je pravdepodobnost vyskytu jadra v zarovnani dlzky L
-
![A[L]=\left\{{\begin{array}{ll}0&{\mbox{ak }}L<w\\p^{w}+\sum _{{i=0}}^{{w-1}}p^{i}(1-p)A[L-i-1]&{\mbox{ak }}L\geq w\\\end{array}}\right.](/vyuka/mbi/images/math/8/a/c/8ac01170bbfa4d0e4666226c83170683.png)
Minimizery: ako usetrit pamat
- k-merom nazveme k za sebou iducich pismen (nukleotidov DNA)
- Zakladne pouzite jadier: pri porovnavani dvoch sekvencii (alebo mnozin sekvencii) uloz vsetky k-mery jednej sekvencie do slovnika (napr. hash tabulky), potom prechadzaj vsetky k-mery druhej sekvencie a hladaj ich v slovniku
- Trik na znizenie potrebnej pamate (napr. program BLAT): ukladaj iba kazdy s-ty k-mer z prvej sekvencie, potom hladaj vsetky k-mery z druhej
- Trochu znizi aj senzitivitu, ale kedze jadra sa klastruju, mame sancu aspon jedno jadro z klastra najst
- Prefikanejsia idea je minimizer: uvazuj vsetky skupiny s po sebe iducich k-merov (sliding window), v kazdej skupine najdi abecedne najmensi k-mer (minimizer) a uloz do slovnika
- Pri posune okna o jedno doprava casto najmensi k-mer zostava ten isty a netreba ho znovu ukladat, cim sa usetri pamat
- Priklad k=5, s=4
AGTGGCTGCCAGGCTGG cGaGGCTGCCaGGtTGG
AGTGG* CGAGG
GTGGC GAGGC
TGGCT AGGCT*
GGCTG GGCTG
GCTGC* GCTGC
CTGCC* CTGCC*
TGCCA TGCCT
GCCAG GCCTG
CCAGG* CCTGG*
CAGGC* CTGGT*
AGGCT* TGGTT
GGCTG GGTTG
GCTGG GTTGG
- Rozdiel je pri hladani: v slovniku nehladame vsetky k-mery druhej sekvencie, ale tiez len minimizery, co moze usetrit cas
- Obzvlast vyhodne ak prva a druha mnozina sekvencii su ta ista, napr. pri hladani prekryvov v citaniach pri skladani genomu. Kazde citanie ma mnozinu minimizerov, ktore sa pouziju ako kluce v slovniku, hodnoty su zoznamy citani. Dvojice citani zdilajuce nejaky minimizer (binning) sa dostanu do jedneho zoznamu a budu uvazovane pri vypocte vzajomneho prekryvu
- V praxi sa do slovnika neuklada lexikograficky najmensi k-mer, ale kazdy k-mer sa prehasuje nejakou funkciou f a zoberie sa ten s minimalnou hodnotou
- Dovod je, ze sa chceme vyhnut, aby minimazermi boli casto sekvencie typu AAAAA, ktore su v biologickych sekvenciach nadreprezentovane a casto nie su funkcne zaujimave
- Minimizery vyuziva napr. aj minimap2, velmi popularny nastroj na zarovnavanie citani navzajom a ku genomom
- Li, Heng. "Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences." Bioinformatics 32.14 (2016): 2103-2110. [1]
- Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. Reducing storage requirements for biological sequence comparison. Bioinformatics. 2004 Dec 12;20(18):3363-9. [2]
MinHash
Odbočka do analýzy web-stránok: Podobnosť textov
Majme množinu webových stránok (webová stránka je postupnosť slov). Chceme nájsť medzi nimi dvojice podobných. Ako môžeme definovať podobnosť dvoch textov?
Jeden zo spôsobov ako to spraviť je pozrieť sa na množstvo slov, spoločných pre jednotlivé dvojice stránok. Očakávame, že čím viac spoločných slov majú, tým sú podobnejšie. Túto mieru podobnosti formalizuje matematický pojem Jaccardovej miery podobnosti.
Nech 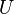 je univerzum slov a nech
je univerzum slov a nech 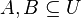 sú jeho podmnožinami (t.j. množiny slov dvoch textov). Potom Jaccardova miera podobnosti
sú jeho podmnožinami (t.j. množiny slov dvoch textov). Potom Jaccardova miera podobnosti 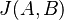 je definovaná nasledovne:
je definovaná nasledovne:
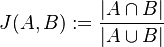
Táto miera nadobúda hodnotu 0 iba v prípade, ak množiny sú disjunktné, a hodnotu 1 iba v prípade, že množiny sú totožné. Inak sa jej hodnota nachádza na otvorenom intervale 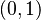 , a čím viac spoločných slov majú, tým je jej hodnota vyššia.
, a čím viac spoločných slov majú, tým je jej hodnota vyššia.
Potom otázku "Ktoré dvojice textov sú podobné?" môžeme preformulovať napríklad ako "Ktoré dvojice textov majú Jaccardovu mieru podobnosti vyššiu ako 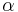 ?", kde
?", kde 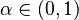 je nejaká prahová hodnota.
je nejaká prahová hodnota.
Exaktný výpočet Jaccardovej miery podobnosti nie je vždy dostatočne rýchly pre účely konkrétnej aplikácie, takže logickým riešením je pokúsiť sa jej hodnotu vypočítať iba približne (t.j. aproximovať).
Aproximácia Jaccardovej miery: MinHash
Nech je daná množina 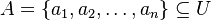 . Nech
. Nech 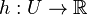 je injektívna náhodná hashovacia funkcia (t.j. bez kolízií). Potom minimálny hash množiny
je injektívna náhodná hashovacia funkcia (t.j. bez kolízií). Potom minimálny hash množiny 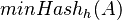 je definovaný nasledovne:
je definovaný nasledovne:
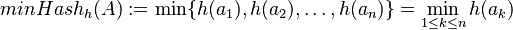
Keďže 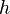 je náhodná hashovacia funkcia, tak sa na hodnotu
je náhodná hashovacia funkcia, tak sa na hodnotu 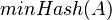 môžeme pozerať ako na náhodnú premennú, ktorá reprezentuje rovnomerne náhodný výber prvku z množiny
môžeme pozerať ako na náhodnú premennú, ktorá reprezentuje rovnomerne náhodný výber prvku z množiny 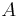 .
.
Nech 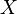 je náhodná premenná, ktorá nadobúda hodnotu 1, ak
je náhodná premenná, ktorá nadobúda hodnotu 1, ak 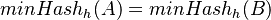 , a inak hodnotu 0. Potom
, a inak hodnotu 0. Potom ![Pr[X=1]=J(A,B)](/vyuka/mbi/images/math/f/8/2/f82bfce9ed488336b83d575fd8bad631.png)
Potom
![E(X)=0\cdot Pr[X=0]+1\cdot Pr[X=1]=Pr[X=1]=J(A,B)](/vyuka/mbi/images/math/4/1/a/41a82418582d44726d6ad464d66c68e5.png) .
.
Z toho vyplýva, že náhodná premenná je nevychýleným odhadom Jaccardovej miery. Je to však veľmi nepohodlný odhad, lebo namiesto celej škály hodnôt od 0 po 1 vracia len dve možnosti.
V štatistike základnou mierou kvality nevychýleného odhadu slúži jeho variancia 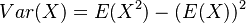 . Spočítajme si postupne obe hodnoty.
. Spočítajme si postupne obe hodnoty.
![E(X^{2})=0^{2}\cdot Pr[X=0]+1^{2}\cdot Pr[X=1]=Pr[X=1]=J(A,B)](/vyuka/mbi/images/math/0/1/2/012881361d10bcbd4d322794b819a493.png)
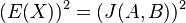
Čiže 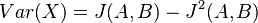 . Aká je maximálna možná hodnota variancie?
. Aká je maximálna možná hodnota variancie?
Táto otázka je ekvivalentná otázke "Aké je maximum funkcie 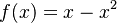 na intervale
na intervale ![[0,1]](/vyuka/mbi/images/math/c/c/f/ccfcd347d0bf65dc77afe01a3306a96b.png) ?". Pre určenie extrémov hladkých funkcií treba nájsť korene jej prvej derivácie. Derivácia tejto funkcie je
?". Pre určenie extrémov hladkých funkcií treba nájsť korene jej prvej derivácie. Derivácia tejto funkcie je 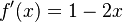 , jej koreň je hodnota
, jej koreň je hodnota 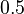 . Hodnota funkcie v tomto bode je rovná
. Hodnota funkcie v tomto bode je rovná 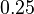 . Čiže
. Čiže 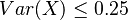 .
.
Ako môžeme tento odhad zlepšiť?
Jedna z možností je zobrať viacero nezávislých hashovacích funkcií 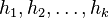 , a spočítať
, a spočítať 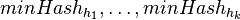 pre obidve množiny. Označme si príslušné náhodné premenné ako
pre obidve množiny. Označme si príslušné náhodné premenné ako 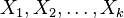 . Každá z nich má strednú hodnotu
. Každá z nich má strednú hodnotu 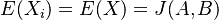 a rovnakú varianciu
a rovnakú varianciu 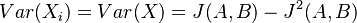 .
.
Nech náhodná premenná 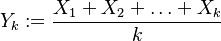 je ich priemer. Potom jej stredná hodnota je rovná
je ich priemer. Potom jej stredná hodnota je rovná ![E(Y_{k})=E\left({\frac {X_{1}+X_{2}+\ldots +X_{k}}{k}}\right)=k^{{-1}}E(X_{1}+X_{2}+\ldots +X_{k})=k^{{-1}}[E(X_{1})+E(X_{2})+\ldots +E(X_{k})]=k^{{-1}}kE(X)=E(X)=J(A,B)](/vyuka/mbi/images/math/a/2/b/a2bd44e6c33c42ef25db86efbbfe2d98.png) . Čiže aj
. Čiže aj 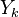 je nevychýleným odhadom Jaccardovej miery.
je nevychýleným odhadom Jaccardovej miery.
Pozrieme sa na jej varianciu: ![Var(Y_{k})=Var\left({\frac {X_{1}+X_{2}+\ldots +X_{k}}{k}}\right)={\dfrac {1}{k^{2}}}Var(X_{1}+X_{2}+\ldots +X_{k}){\overset {*}{=}}{\dfrac {1}{k^{2}}}[Var(X_{1})+\ldots Var(X_{k})]={\dfrac {1}{k^{2}}}k\cdot Var(X)={\dfrac {Var(X)}{k}}\leq {\dfrac {1}{4k}}](/vyuka/mbi/images/math/7/c/2/7c213a4c85784d2ff294349150587e79.png)
(*) tento prechod je možný len kvôli tomu, že premenné 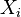 sú nezávislé.
sú nezávislé.
Vidíme teda, že varianciu (resp. kvalitu) môžeme potlačiť na ľubovoľne malú postupným zvýšením počtu hashov.
Všimnite si, že premenná 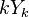 (t.j. nie priemer, ale súčet jednotlivých ) je súčtom nezávislých indikátorov s rovnakou distribúciou, a teda má binomické rozdelenie s parametrami
(t.j. nie priemer, ale súčet jednotlivých ) je súčtom nezávislých indikátorov s rovnakou distribúciou, a teda má binomické rozdelenie s parametrami 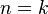 a
a 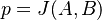 .
.
Druhá možnosť zlepšenia je nechať jednu hashovaciu funkciu, ale porovnávať nie 1, ale k najmenších hashov dvoch množín. Vedie to ku podobnému asymptotickému správaniu.
Návrat do porovnávania sekvencií
Ako "slová" použijeme všetky súvislé podreťazce fixnej dĺžky (dĺžka sa tradične označuje ako 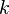 ) danej sekvencie. Tieto podreťazce sa tradične označujú ako -mery.
Potom na hľadanie dvoch podobných čítaní z množiny čítaní môžeme použiť minhash.
) danej sekvencie. Tieto podreťazce sa tradične označujú ako -mery.
Potom na hľadanie dvoch podobných čítaní z množiny čítaní môžeme použiť minhash.
- Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome biology. 2016 Dec;17(1):1-4. [3]