CB07
Z MBI
Obsah
[skryť]E-hodnota (E-value) zarovnania
- Priklady k tejto casti v prezentacii pdf
- Mame dotaz dlzky m, databazu dlzky n, skore najlepsieho lokálneho zarovnania S
- E-value je ocakavany pocet zarovnani so skore aspon S ak dotaz aj databaza su nahodne
- Hrackarsky priklad: dotaz dlzky m=10, databaza dlzky n=300, S=6
- Zoberme nas nahodny model s obsahom GC 50%
- Mame vrece s gulockami oznacenymi A,C,G,T, z kazdej 25%
- Vytiahneme gulicku, zapiseme si pismeno, hodime ju naspat, zamiesame a opakujeme s dalsim pismenom atd az kym nevygenerujeme m pismen pre dotaz a n pismen pre databazu
- Pre nase vygenerovane sekvencie spocitame, kolkokrat sa dotaz vyskytuje v databaze
- Cely experiment opakujeme vela krat a spocitame priemerny pocet vyskytov, co bude odhad E-value
Vypocet strednej hodnoty vzorcom namiesto simulacie (rychlejsie)
- zlozita matematicka teoria [1]
- E-value sa priblizne da odhadnut vzorcom:
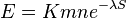
- n a m su dlzky porovnavanych sekvencii, S je skore, K a lambda su parametre, ktore zavisia od skorovacej schemy a od frekvencii vyskytu jednotlivych baz v nasom modeli nahodnej sekvencie.
- Napr blastn pre skorovaci system zhoda 1, nezhoda -1, medzera -2 používa lambda=0.800, K=0.0640
-
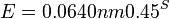 .
.
- Zdvojnásobenie dĺžky databázy alebo dĺžky dotazu zdvojnásobí E-value
- Zníženie skóre o 1 tiež zhruba zdvojnásobí E-value (delenie 0.45, t.j. nasobenie 2.2)
- Cislo, ktorym nasobime, zalezi od lambda a teda od skorovacej schemy a frekvencii vyskytu baz
Praktická ukážka tvorby stromov
- V UCSC browseri mozeme ziskavat viacnasobne zarovnania jednotlivych genov (nukleotidy alebo proteiny). Nasledujuci postup nemusite robit, subor si stiahnite tu: http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb06/cb06-aln.fa
- UCSC browseri si pozrieme usek ludskeho genomu (verzia hg38) chr6:135,851,998-136,191,840 s genom PDE7B (phosphodiesterase 7B) [2]
- Na modrej liste zvolime Tools, Table browser. V nastaveniach tabuliek Group: Genes and Gene Predictions, Track: GENCODE v 32., zaklikneme Region: position, a Output fomat: CDS FASTA alignment a stlacime Get output
- Na dalsej obrazovke zaklikneme show nucleotides, zvolime MAF table multiz100way a vyberieme si, ktore organizmy chceme. V nasom pripade z primatov zvolime chimp, rhesus, bushbaby, z inych cicavcov mouse, rat, rabbit, pig, cow, dog, elephant a z dalsich organizmov opposum, platypus, chicken, stlacime Get output.
- Vystup ulozime do suboru, nechame si iba prvu formu genu (ENST00000308191.11_hg38), z mien sekvencii zmazeme spolocny zaciatok (ENST00000308191.11_hg38), pripadne celkovo prepiseme mena na anglicke nazvy
- Skusme zostavit strom na stranke http://www.ebi.ac.uk/Tools/phylogeny/clustalw2_phylogeny/
- Distance correction: ako na prednáške, z počtu pozorovaných mutácií na evolučný čas
- Exclude gaps: vynechať všetky stĺpce s pomlčkami
- Clustering method: UPGMA predpokladá molekulárne hodiny, spájanie susedov nie
- P.I.M. vypíš aj maticu vzdialeností (% identity, pred korekciou)
- Vo vyslednom strome by sme mali zmenit zakorenenie, aby sme mali sliepku (chicken) ako outgroup
- Výsledky z programu http://www.phylogeny.fr/alacarte.cgi , ktorý podporuje aj bootstrap:
- Vysledok s povodnym zakorenenim
- Vysledok so spravnym zakorenenim (chicken = outgroup)
- "Spravny strom" [3] v nastaveniach Conservation track-u v UCSC browseri (podla clanku Murphy WJ et al Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001 [4])
- Nas strom ma dost zlych hran: zle postavenie hlodavcov, ale aj slona a psa. Zle postavenie hlodavcov môže byť spôsobené long branch attraction.
- Ak chcete skusit zostavit aj zarovnania, treba zacat z nezarovnanych sekvencii: [5]
![[3]](http://genome-euro.ucsc.edu/images/phylo/hg38_100way.png){kind=link}
Ďalšia zaujímavá webstránka s veľa nástrojmi https://usegalaxy.eu/
- na tvorbu stromov sa dá použiť IQ-TREE
- modely vid tu: https://github.com/Cibiv/IQ-TREE/wiki/Substitution-Models
- vysledok #CB:phylo
- viewer napr. http://phylotree.hyphy.org/
Gény, evolúcia a komparatívna genomika v UCSC genome browseri (cvičenie pri počítači)
- Zobrazme si gén CLCA4 [6]
- Zapnite si štandardnú sadu track-ov
- Po kliknutí na gén si môžete prečítať o jeho funkcii, po kliknutí na ľavú lištu alebo na názov tracku v zozname na spodku stránky si môžete prečítať viac o tracku a meniť nastavenia
- V tracku RefSeq genes si všimnite, že v tejto databáze má tento gén dve formy zostrihu, jedna z nich sa považuje za nekódujúcu, pretína sa aj s necharakterizovanou nekódujúcou RNA na opačnom vlákne
- Track RefSeq a jeho subtrack RefSeq Curated treba zapnut na pack
- Nižšie vidíte track H3K27Ac Mark (Often Found Near Regulatory Elements) on 7 cell lines from ENCODE, kde bola táto histónová modifikácia v okolí génu detegovaná?
- Všimnite si aj track DNase I Hypersensitivity, ktorý zobrazuje otvorený chromatin, prístupný pre viazanie transkripčných faktorov. Všimnite si jeho súvis s H3K27Ac trackom
- Obidva tracky sú súčasťou tracku ENCODE regulation, v ktorom si môžete zapnúť aj ďalšie pod-tracky
- Vsimnime si track Vertebrate Multiz Alignment & Conservation (100 Species)
- v spodnej casti tracku vidime zarovnania s roznymi inymi genomami
- v nastaveniach tracku zapnite Element Conservation (phastCons) na full a Conserved Elements na dense
- v tomto tracku vidíme PhyloP, co zobrazuje uroven konzerovanosti danej bazy len na zaklade jedneho stlpca zarovnania a dva vysledky z phyloHMM phastCons, ktory berie do uvahy aj okolite stlpce
- Konkretne cast Conserved elements zobrazuje konkretne useky, ktore su najvac konzervovane
- Ak chceme zistit, kolko percent genomu tieto useky pokryvaju, ideme na modrej liste do casti Tools->Table browser, zvolime group Comparative genomics, track Conservation, table 100 Vert. El, region zvolime genome (v celom genome) a stlacime tlacidlo Summary/statistics, dostaneme nieco taketo:
| item count | 10,350,729 |
| item bases | 162,179,256 (5.32%) |
| item total | 162,179,256 (5.32%) |
| smallest item | 1 |
| average item | 16 |
| biggest item | 3,732 |
| smallest score | 186 |
| average score | 333 |
| biggest score | 1,000 |
- Ak by nas zaujimali iba velmi dlhe "conserved elements", v Table browser stlacime tlacidlo Filter a na dalsej obrazovke do policka Free-form query dame chromEnd-chromStart>=1500
- Potom mozeme skusit Summary/Statistics alebo vystup typu Hyperlinks to genome browser a Get output - dostaneme zoznam tychto elementov a kazdy si mozeme jednym klikom pozriet v browseri, napr. taketo
- Pozrime si teraz ten isty gen CLCA4 v starsej verzii genomu hg18 [7]
- V casti Genes and Gene Prediction Tracks zapnite track Pos Sel Genes, ktory obsahuje geny s pozitivnym vyberom (cervenou, pripadne slabsie fialovou a modrou)
- Ked kliknete na cerveny obdlznik pre tento gen, uvidite, v ktorych castiach fylogenetickeho stromu bol detegovany pozitivny vyber
- Po priblizeni do jedneho z exonov [8] vidite dosledky nesynonymnych mutacii
Poznamka: Existuju aj webservery na predikciu pozitivneho vyberu, napriklad tieto dva:
- Selecton, clanok
- Data monkey clanok
- Skusili sme na Selecton poslat CLCA4 zo 7 cicavcov, subor tu: [9]
Objavenie génu HAR1 pomocou komparatívnej genomiky
- Pollard KS, Salama SR, Lambert N, et al. (September 2006). "An RNA gene expressed during cortical development evolved rapidly in humans". Nature 443 (7108): 167–72. doi:10.1038/nature05113. PMID 16915236. pdf
- Zobrali všetky regióny dĺžky aspoň 100bp s > 96% podobnosťou medzi šimpanzom a myšou/potkanom (35,000)
- Porovnali s ostatnými cicavcami, zistili, ktoré majú veľa mutáci v človeku, ale málo inde (pravdepodobnostný model)
- 49 štatisticky významných regiónov, 96% nekódujúcich oblastiach
- Najvýznamnejší HAR1: 118nt, 18 substitúcii u človeka, očakávali by sme 0.27. Iba 2 zmeny medzi šimpanzom a sliepkou (310 miliónov rokov), ale nebol nájdený v rybách a žabe.
- Nezdá sa byť polymorfný u človeka
- Prekrývajúce sa RNA gény HAR1A a HAR1B
- HAR1A je exprimovaný v neokortexe u 7 a 9 týždenných embrií, neskôr aj v iných častiach mozgu (u človeka aj iných primátov)
- Všetky substitúcie v človeku A/T->C/G, stabilnejšia RNA štruktúra (ale tiež sú blízko k telomére, kde je viacej takýchto mutácii kvôli rekombinácii a biased gene conversion)
Cvičenie pri počítači
- Môžete si pozrieť tento region v browseri: chr20:63102114-63102274 (hg38), pricom ak sa este priblizite, uvidite zarovnanie aj s bazami a mozete vidiet, ze vela zmien je specifickych pre cloveka
Hľadanie génov
K hladaniu genov pozri aj prezentacie pdf
Hľadanie génov v prokaryotických genómoch
- ORF: open reading frame, jednoduche hladanie
- ako najst zaciatok, ako rozlisit psedogeny a nahodne ORF-y
- samotrenujuce sa HMM, codon bias, GC%
E. coli http://nar.oxfordjournals.org/content/34/1/1.full
- Prvykrat sekvenovana a anotovana 1997
- Porovnanie s verziou 2005 (oprava sekvenovacích chýb aj chýb v anotácii)
- 682 zmien v start kodone
- 31 génov zrušených
- 48 nových génov
- Celkovo asi 4464 génov
Programy na anotovanie prokaryotických genómov
Histónové modifikácie
- A. Barski, S. Cuddapah, K. Cui, T. Roh, D. Schones, Z. Wang, G. Wei, I. Chepelev, K. Zhao (2007) High-Resolution Profiling of Histone Methylations in the Human Genome Cell, Volume 129, Issue 4, Pages 823-837 pdf
Gény v ľudskom genóme
- What is a gene, post-ENCODE? History and updated definition. Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, Emanuelsson O, Zhang ZD, Weissman S, Snyder M.
- Most "dark matter" transcripts are associated with known genes. H Van Bakel, C Nislow, BJ Blencowe, TR Hughes - PLoS Biol, 2010
- Transcribed dark matter: meaning or myth? CP Ponting, TG Belgard - Human molecular genetics, 2010
- Landscape of transcription in human cells. Djebali et al (ENCODE), Nature 2012