MBI 2016/2017
Na tejto stránke nájdete orientačný rozvrh semestra, ktorý budeme priebežne aktualizovať. Poznámky z prednášok a ďalšie rozširujúce materiály budú pribúdať po skončení príslušných prednášok.
Literatúra:
- BV: Brejová, Vinař: Metódy v bioinformatike. (predbežná verzia skrípt, iba niektoré prednášky)
- Verzia z 1.10.2015: pdf
- DEKM: Durbin, Eddy, Krogh, Mitchison: Biological sequence analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press 1998.
- ZB: Zvelebil, Baum: Understanding Bioinformatics. Taylor & Francis 2008.
Pri prednáškach uvádzame kapitoly najviac pokrývajúce učivo, ktoré plánujeme prebrať. Prezentácia materiálu v rámci prednášok sa obvykle nezhoduje s prezentáciou v učebniciach. Uvedené kapitoly by mali hlavne slúžiť ako doplňujúci materiál pre samoštúdium.
PR: spoločná prednáška, CI: cvičenia pre informatikov, CB: cvičenia pre biológov
Prezentácie
- všetky prednášky v jednom pdf
- všetky cvičenia pre biológov v jednom pdf
- všetky cvičenia pre informatikov v jednom pdf
| Týždeň 19.-25.9. | |||
| PR: | Úvod, administratíva, sekvenovanie a zostavovanie genómov | ||
| BV kap. 1 | |||
| CI: | Úvod do biológie | poznámky | |
| ZB kap. 1 | |||
| CB: | Úvod do informatiky, UCSC genome browser | poznámky | |
| Týždeň 26.9.-2.10 | |||
| PR: | nebude, cvičenia pre biológov začínajú 15:40 v F1-328 | ||
| CI: | Úvod do pravdepodobnosti, pokrytie pri sekvenovaní | poznámky | |
| CB: | Úvod do dynamického programovania, úvod do pravdepodobnosti | poznámky | |
| Týždeň 3.-9.10. | |||
| PR: | Zarovnávanie sekvencií: Smith-Waterman, Needleman-Wunsch, skórovanie | ||
| BV kap. 2, DEKM kap. 2.1-2.4, 2.8, ZB kap. 4.1-4.4, 5.1-5.2 | |||
| CI: | Úvod do dynamického programovania, proteomika | poznámky | |
| CB: | Dynamické programovanie pre zarovnávanie sekvencií, dotploty | poznámky | |
| Týždeň 10.-16.10. | |||
| PR: | Zarovnávanie sekvencií: heuristické zarovnávanie (BLAST), štatistická významnosť zarovnaní, zarovnávanie genómov, viacnásobné zarovnanie | ||
| BV kap. 2, DEKM kap. 2.5, 2.7, 6.1-6.3; ZB kap. 4.5-4.7, 5.3-5.5 | |||
| CI: | Pokročilé algoritmy pre zarovnávanie sekvencií | poznámky | |
| CB: | Programy na zarovnávanie sekvencií, dotploty, skórovacie schémy | poznámky | |
| Týždeň 17.-23.10. | |||
| PR: | Hľadanie génov, skryté Markovove modely | ||
| BV kap. 4, DEKM kap. 3; ZB kap. 9.3, 10.4-10.7 | |||
| CI: | BLAST, jadrá s medzerami | poznámky | |
| CB: | Skryté Markovove modely, E-value, hľadanie génov | poznámky | |
| Týždeň 24.10.-30.10. | |||
| PR: | Rekonštrukcia fylogenetických stromov (úspornosť, metóda spájania susedov, modely evolúcie) | ||
| BV kap. 3, DEKM kap. 7,8; ZB kap. 7, 8.1-8.2 | |||
| CI: | Algoritmy pre HMM | poznámky | |
| CB: | Substitučné modely, bootstrap, praktická ukážka tvorby stromov | poznámky | |
| Týždeň 31.10-6.11. | |||
| PR: | Komparatívna genomika, detekcia pozitívneho a purifikačného výberu, komparatívne hľadanie génov, fylogenetické HMM | ||
| BV kap. 5, ZB kap. 9.8, 10.8 | |||
| CI: | Substitučné modely | poznámky | |
| CB: | Gény v UCSC browseri, komparatívna genomika, Fitchov aloritmus | poznámky | |
| Týždeň 7.-13.11. | |||
| PR: | Expresia génov, zhlukovanie, klasifikácia, regulačné siete, transkripčné faktory, motívy v sekvenciách | ||
| DEKM kap. 5.1, 11.5, ZB kap. 6.6,15.1,16.1-16.5,17.1 | |||
| CI: | Felsensteinov algoritmus, algoritmy pre HMM a phyloHMM | poznámky | |
| CB: | Zhlukovanie algoritmom k-means, nadreprezentácia, multiple testing correction | poznámky | |
| Týždeň 14.-20.11. sviatok | |||
| Týždeň 21.-27.11. | |||
| PR: | Štruktúra a funkcia proteínov | ||
| DEKM kap. 5; ZB kap. 4.8-4.10, 6.1-6.2, 13.1-13.2 | |||
| CI: | Hľadanie motívov EM a Gibbsovým vzorkovaním | poznámky | |
| CB: | Úvod do bezkontextových gramatík, microarray dáta, Uniprot, PSI-BLAST, Pfam | poznámky | |
| Týždeň 28.11.-4.12. | |||
| PR: | RNA, sekundárna štruktúra, Nussinovovej algoritmus, stochastické bezkontextové gramatiky, profily RNA rodín | ||
| DEKM kap. 10, ZB kap. 11.9 | |||
| CI: | Protein threading pomocou celočíselného lineárneho programovania, zhlukovanie algoritmom k-means, úvod do bezkontextových gramatík | poznámky | |
| CB: | RNA štruktúra, MEME, transkripčné faktory v SGD | poznámky | |
| Týždeň 5.-11.12. | |||
| PR: | Populačná genetika | ||
| CI: | RNA štruktúra, ukážky biologických databáz | poznámky | |
| CB: | Zhrnutie semestra, grafy, populačná genetika | poznámky | |
| Týždeň 12.-18.12. | |||
| CI: | Zhrnutie semestra, štruktúra populácie | poznámky | |
| PR: | Nepovinné prezentácie | ||
Obsah
- 1 CI01
- 2 CB01
- 3 CI02
- 4 CB02
- 5 CI03
- 6 CB03
- 7 CI04
- 8 CB04
- 9 CI05
- 10 CB05
- 11 CI06
- 12 CB06
- 13 CI07
- 14 CB07
- 15 CI08
- 16 CB08
- 17 CI09
- 18 CB09
- 19 CI10
- 20 CB10
- 21 CI11
- 22 CB11
- 23 CI12
CI01
Úvod do biológie pre informatikov
Užitočné linky
CB01
Používanie počítačov v M 217
- V textovom menu pri štarte zvoľte Linux, v prihlasovacom menu zadajte užívatela bioinf, heslo dostanete
- Na dolnom okraji obrazovky je lišta s často používanými nástrojmi, napr. internetový prehliadač Firefox
- Vo Firefoxe si otvorte stránku predmetu http://compbio.fmph.uniba.sk/vyuka/mbi/ čast Prednášky a poznámky, nalistujte materiály k dnešnému cvičeniu
UCSC genome browser
- On-line grafický nástroj na prezeranie genómov
- Konfigurovateľný, veľa možností, ale pomerne málo organizmov
- V programe Firefox choďte na stránku UCSC genome browser http://genome-euro.ucsc.edu/ (európsky mirror stránky http://genome.ucsc.edu/ )
- Hore v modrom menu zvoľte Genomes, potom zvoľte ľudský genóm verzia hg38. Do okienka search term zadajte HOXA2. Vo výsledkoch hľadania (Known genes) zvoľte gén homeobox A2 na chromozóme 7.
- Pozrime si spolu túto stránku
- V hornej časti sú ovládacie prvky na pohyb vľavo, vpravo, približovanie, vzďaľovanie
- Pod tým schéma chromozómu, červeným vyznačená zobrazená oblasť
- Pod tým obrázok vybranej oblasti, rôzne tracky
- Pod tým zoznam všetkých trackov, dajú sa zapínať, vypínať a konfigurovať
- Po kliknutí na obrázok sa často zobrazí ďalšia informácia o danom géne alebo inom zdroji dát
- V génoch exony hrubé, UTR tenšie, intróny vodorovné čiary
- Koľko má HOXA2 exónov? Na ktorom chromozóme a pozícii je? Pozor, je na opačnom vlákne. Ako je táto skutočnosť naznačená na obrázku?
- V tracku GENCODE kliknite na gén, mali by ste sa dostať na stránku popisujúcu jeho rôzne vlastnosti. Čo ste sa dozvedeli o jeho funkcii?
- Na tejto stránke nájdite linku na stiahnutie proteínovej sekvencie. Aké sú prvé štyri aminokyseliny?
Sekvenovanie v UCSC genome browseri
- Vráťte sa na UCSC genome browser http://genome.ucsc.edu/
- Pozrieme si niekoľko vecí týkajúcich sa sekvenovania a skladania genómov
- Hore v modrom menu zvoľte Genomes, časť Other
- Na ďalšej stránke zvoľte človeka a pomocou menu Human Assembly zistite, kedy boli pridané posledné dve verzie ľudského genómu (hg19 a hg38)
- Na tej istej stránke dole nájdete stručný popis zvolenej verzie genómu. Pre ktoré oblasti genómu máme v hg38 najviac alternatívnych verzií? (haplotypes)
- Prejdite na región chr21:31,250,000-31,300,000 v hg19 touto linkou: [1]
- Zapnite si tracky Mapability a RepeatMasker na "full"
- Mapability: nakoľko sa daný úsek opakuje v genóme a či teda vieme jednoznačne jeho čítania namapovať pri použití Next generation sequencing
- Ako a prečo sa pri rôznych dĺžkach readov líšia? (Keď kliknete na linku "Mapability", môžete si prečítať bližšie detaily.)
- Približne v strede zobrazeného regiónu je pokles mapovateľnosti. Akému typu opakovania zodpovedá? (pozrite track RepeatMasker)
- Zapnite si tracky "Assembly" a "Gaps" a pozrite si región chr2:110,000,000-110,300,000 v hg19: [2] Aká dlhá je neosekvenovaná medzera (gap) v strede tohto regiónu? Približnú veľkosť môžete odčítať z obrázku, presnejší údaj zistíte kliknutím na čierny obdĺžnik zodpovedajúci tejto medzere (úplne presná dĺžka aj tak nebola známa, nakoľko nebola osekvenovaná).
- Cez menu položku View, In other genomes si pozrite, ako zobrazený úsek vyzerá vo verzii hg38. Ako sa zmenila dĺžka z pôvodných 300kb?
- Prejdite na genóm Rhesus, verzia rheMac2, región chr7:59,022,000-59,024,000 [3], zapnite si tracky Contigs, Gaps, Quality scores
- Aké typy problémov v kvalite sekvencie v tomto regióne vidíte?
- Opäť si môžete pozrieť, či sa problémy odstránili a ako sa zmenila dĺžka sekvencie v najnovšej verzii rheMac8
CI02
Úvod do pravdepodobnosti
- Myšlienkový experiment, v ktorom vystupuje náhoda, napr. hod ideálnou kockou/mincou
- Výsledkom experimentu je nejaká hodnota (napr. číslo, alebo aj niekoľko čísel, reťazec)
- Túto neznámu hodnotu budeme volať náhodná premenná
- Zaujíma nás pravdepodobnosť, s akou náhodná premenná nadobúda jednotlivé možné hodnoty
- T.j. ak experiment opakujeme veľa krát, ako často uvidíme nejaký výsledok
Príklad 1: hodíme idealizovanou kockou, premenná X bude hodnota, ktorú dostaneme
- Možné hodnoty 1,2,..,6, každá rovnako pravdepodobná
- Pišeme napr. Pr(X=2)=1/6
Príklad 2: hodíme 2x kockou, náhodná premenná X bude súčet hodnôt, ktoré dostaneme
- Možné hodnoty: 2,3,...,12
- Každá dvojica hodnôt (1,1), (1,2),...,(6,6) na kocke rovnako pravdepodobná, t.j. pravdepodobnosť 1/36
- Súčet 5 môžeme dostať 1+4,2+3,3+2,4+1 - t.j. P(X=5) = 4/36
- Súčet 11 môžeme dostať 5+6 alebo 6+5, t.j. P(X=11) = 2/36
- Rozdelenie pravdepodobnosti: (tabuľka udávajúca pravdepodobnosť pre každú možnú hodnotu)
hodnota i: 2 3 4 5 6 7 8 9 10 11 12 Pr(X=i): 1/36 2/36 3/36 4/36 5/36 6/36 5/36 4/36 3/36 2/36 1/36
- Overte, ze súčet pravdepodobností je 1
Stredná hodnota E(X):
- priemer z možných hodnôt váhovaných ich pravdepodobnosťami
- v našom príklade
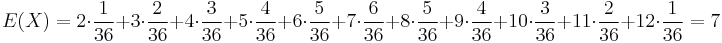
- Ak by sme experiment opakovali veľa krát a zrátali priemer hodnôt X, ktoré nám vyšli, dostali by sme číslo blízke E(X)
- Iný výpočet strednej hodnoty:
- X=X1+X2, kde X1 je hodnota na prvej kocke a X2 je hodnota na druhej kocke
-
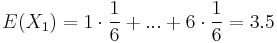 , podobne aj E(X2) = 3.5
, podobne aj E(X2) = 3.5
- Platí, že E(X1+X2)=E(X1) + E(X2) a teda E(X) = 3.5 + 3.5 = 7
- Pozor, pre súčin a iné funkcie takéto vzťahy platiť nemusia, napr.
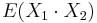 nie je vždy
nie je vždy 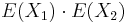
Počítanie pokrytia genómov
- Pozrite tiez grafy k pravdepodobnosti: pdf
- Nas problem: spocitanie pokrytia
- G = dlzka genomu, napr. 1 000 000 (predpokladajme, že je cirkulárny)
- N = pocet čítaní (readov), napr. 10 000
- L = dlzka readu, napr. 1000
- Celkova dlzka čítaní NL, pokrytie (coverage) NL/G, v nasom pripade 10x
- V priemere kazda baza pokryta 10x
- Niektore su ale pokryte viackrat, ine menej.
- Zaujimaju nas otazky typu: kolko baz ocakavame, ze bude pokrytych menej ako 3x?
- Dolezite pri planovani experimentov (ake velke pokrytie potrebujem na dosiahnutie urcitej kvality)
- Pokrytie genomu: predpokladame, ze kazde čítanie zacina na nahodnej pozicii zo vsetkych moznych G
- Takze ak premenna Y_i bude zaciatok i-teho čítania, jej rozdelenie bude rovnomerne
- P(Y_i=1) = P(Y_i=2) = ... = P(Y_i=G) = 1/G
- Aka je pravdepodobnost ze nejake konkretne i-te čítanie pokryva konkretnu poziciu j?
- P(Y_i>=j-L+1 and Y_i<=j) = P(Y_i=j-L+1)+...+P(Y_i=j) = L/G, oznacme tuto hodnotu p, nasom priklade p=0.001 (1 promile)
- Uvazujme premennu X_j, ktora udava pocet čítaní pokryvajucich poziciu j
- mozne hodnoty 0..N
- i-te čítanie pretina poziciu j s pravdepodobnostou p=L/G
- to iste ako keby sme N krat hodili mincou, na ktorej spadne hlava s pravd. p a znak 1-p a oznacili ako X_j pocet hlav
- Priklad: majme mincu, ktora ma hlavu s pr. 1/4 a hodime ju 3x.
HHH 1/64 HHT 3/64 HTH 3/64 HTT 9/64 THH 3/64 THT 9/64 TTH 9/64 TTT 27/64
- P(X_j=3) = 1/64, P(X_j=2)=9/64, P(X_j=1)=27/64, P(X_j=0)=27/64
- taketo rozdelenie pravdepodobnosti sa vola binomicke
- P(X_j = k) = (N choose k) p^k (1-p)^(N-k), kde
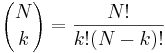 a n! = 1*2*...*n
a n! = 1*2*...*n
- napr pre priklad s troma hodmi kockou P(X_j=2) = 3!/(2!*1!) * (1/4)^2 * (3/4)^1 = 9/64
- Zle sa pocita pre velke N, preto sa niekedy pouziva aproximacia Poissonovym rozdelenim s parametrom lambda = Np, ktore ma
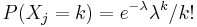
- Spat k sekvenovaniu: vieme spocitat rozdelenie pravdepodobnosti a tiez napr. P(X_i<3) = P(X_i=0)+P(X_i=1)+P(X_i=2) = 0.000045+0.00045+0.0023=0.0028 (v priemere ocakavame 45 baz nepokrytych, 2800 pokrytých menej ako 3 krát)
- Takyto graf, odhad, vieme lahko spravit pre rozne pocty čítaní a tak naplanovat, kolko čítaní potrebujeme
Chceme tiež odhadnúť počet kontigov (podľa článku E.S. Lander and M.S. Waterman. "Genomic mapping by fingerprinting random clones: a mathematical analysis." Genomics 2.3 (1988): 231-239 [4])
- Ak niekoľko báz vôbec nie je pokrytých čítaniami, preruší sa kontig
- Vieme, koľko báz je v priemere nepokrytých, ale niektoré môžu byť vedľa seba
- Nový kontig vznikne aj ak sa susedné čítania málo prekrývajú
- Predpokladajme, že na spojenie dvoch čítaní potrebujeme prekryv aspoň T=50
- Nech p je pravdepodobnosť, ze dané čítanie i bude posledné v kontigu
- Aby sa to stalo, žiadne čítanie j!=i nesmie začínať v prvých L-T bázach kontigu i
- Každé čítanie tam začína s pravdepodobnosťou q=(L-T)/G
- Ak X je počet čítaní, ktoré zacinaju v tomto useku, tak p = Pr(X=0) = (1-q)^(N-1) podla binomickeho rozdelenia
- v priemere ich tam zacne E(X) = (N-1)(L-T)/G co je zhruba N(L-T)/G
- Jednoduchší vzorec pre p dostaneme ak binomické rozdelenie premennej X aproximujeme Poissonovým s parametrom
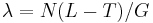 (t.j. aby mali rovnakú strednú hodnotu)
(t.j. aby mali rovnakú strednú hodnotu)
- V Poissonovom rozdelení p = Pr(X=0) = exp(-lambda) = exp(-N(L-T)/G)
- Presnosť aproximácie: pre parametre N,L,G,T uvedené vyššie dostaneme z binomického rozdelenia p=7.459e-5, z Poissonovho 7.485e-5
- Pre N čítaní dostaneme priemerný počet kontigov N*p = N*exp(-N(L-T)/G)
- NL/G je pokrytie, N(L-T)/G je pokrytie, ak by sme dĺžku každého čítania skrátili o dĺžku prekryvu
- Pre T=50 dostaneme priemerný počet koncov kontigov 0.75 (ak pokryjeme celý kruh, máme nula koncov, preto je hodnota menšia ako 1). Ak znížime N na 5000 (5x pokrytie) dostaneme 43 kontigov
- Môže sa zdať zvláštne, ze pri priemernom pocte nepokrytych baz 45 mame pocet koncov v priemere menej ako jedna. Situacia je vsak taka, ze pri opakovaniach tohto experimentu casto dostavame jeden suvisly kontig, ale ak je uz aspon jeden koniec kontigu, byva tam pomerne velka medzera. Tu je napriklad 50 opakovani expertimentu s T=0, priemerny pocet koncov je 0.55, priemerny pocet nepokrytych baz je 49.
nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 274 koncov: 2 nepokr: 282 koncov: 1 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 8 koncov: 1 nepokr: 0 koncov: 0 nepokr: 12 koncov: 1 nepokr: 0 koncov: 0 nepokr: 122 koncov: 1 nepokr: 135 koncov: 1 nepokr: 111 koncov: 1 nepokr: 13 koncov: 1 nepokr: 1 koncov: 1 nepokr: 56 koncov: 1 nepokr: 265 koncov: 1 nepokr: 0 koncov: 0 nepokr: 10 koncov: 1 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 130 koncov: 1 nepokr: 217 koncov: 1 nepokr: 3 koncov: 1 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 86 koncov: 1 nepokr: 139 koncov: 2 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 76 koncov: 1 nepokr: 221 koncov: 1 nepokr: 26 koncov: 1 nepokr: 0 koncov: 0 nepokr: 1 koncov: 1 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 0 koncov: 0 nepokr: 12 koncov: 1 nepokr: 103 koncov: 2 nepokr: 0 koncov: 0 nepokr: 71 koncov: 1 nepokr: 69 koncov: 1 nepokr: 0 koncov: 0
- Tento jednoduchy model nepokryva vsetky faktory:
- čítania nemaju rovnaku dlzku
- Problemy v zostavovani kvoli chybam, opakovaniam a pod.
- čítania nie su rozlozene rovnomerne (cloning bias a pod.)
- Vplyv koncov chromozomov pri linearnych chromozomoch
- Uzitocny ako hruby odhad
- Na spresnenie mozeme skusat spravit zlozitejsie modely, alebo simulovat data
- Poznamka: pravdepodobnosti z binomickeho rozdelenia mozeme lahko spocitat napr. statistickym softverom R. Tu su prikazy, ktore sa na to hodia, pre pripad, ze by vas to zaujimalo:
dbinom(10,1e4,0.001); #(12.5% miest ma pokrytie presne 10) pbinom(10,1e4,0.001,lower.tail=TRUE); #(58% miest ma pokrytie najviac 10) dbinom(0:30,1e4,0.001); #tabulka pravdepodobnosti [1] 4.517335e-05 4.521856e-04 2.262965e-03 7.549258e-03 1.888637e-02 [6] 3.779542e-02 6.302390e-02 9.007019e-02 1.126216e-01 1.251601e-01 [11] 1.251726e-01 1.137933e-01 9.481826e-02 7.292252e-02 5.207187e-02 [16] 3.470068e-02 2.167707e-02 1.274356e-02 7.074795e-03 3.720595e-03 [21] 1.858621e-03 8.841718e-04 4.014538e-04 1.743354e-04 7.254524e-05 [26] 2.897743e-05 1.112843e-05 4.115040e-06 1.467156e-06 5.050044e-07 [31] 1.680146e-07
Zhrnutie
- Pravdepobnostny model: myslienkovy experiment, v ktorom vystupuje nahoda, napr. hod idealizovanou kockou
- Vysledok je hodnota, ktoru budeme volat nahodna premenna
- Tabulka, ktora pre kazdu moznu hodnotu nahodnej premennej urci jej pravdepodobnost, sa vola rozdelenie pravdepodobnosti, sucet hodnot v tabulke je 1
- Znacenie typu P(X=7)=0.1
- Priklad: mame genom dlzky G=1mil., nahodne umiestnime N=10000 čítaní dlzky L=1000
- Nahodna premenna X_i je pocet čítaní pokryvajucich urcitu poziciu i
- Podobne, ako keby sme N krat hodili kocku, ktora ma cca 1 promile sancu padnu ako hlava a 99.9% ako znak a pytame sa, kolko krat padne znak (1 promile sme dostali po zaukruhleni z L/(G-L+1))
- Rozdelenie pravdepobnosti sa v tomto pripade vola binomicke a existuje vzorec, ako ho spocitat
- Takyto model nam moze pomoct urcit, kolko čítaní potrebujeme osekvenovat, aby napr. aspon 95% pozicii bolo pokrytych aspon 4 čítaniami
CB02
Dynamické programovanie
- Túto techniku uvidíme na ďalšej prednáške na hľadanie zarovnaní (alignments)
- Uvažujme problém platenia pomocou najmenšieho počtu mincí
- Napr. máme mince hodnoty 1,2,5 centov, z každej dostatok kusov
- Ako môžeme zaplatiť určitú sumu, napr. 13 centov, s čo najmenším počtom mincí?
- Aké je riešenie? 5+5+2+1 (4 mince)
- Všeobecná formulácia:
- Vstup: hodnoty k mincí m_1,m_2,...,m_k a cieľová suma X (všetko kladné celé čísla)
- Výstup: najmenší počet mincí, ktoré potrebujeme na zaplatenie X
- V našom príklade k=3, m_1 = 1, m_2 = 2, m_3 = 5, X=13
- Jednoduchý spôsob riešenia: použi najväčšiu mincu, ktorá je najviac X, odčítaj od X, opakuj
- Príklad: najprv použijeme mincu 5, zostane X=8, použijeme opäť mincu 5, zostane X=3, použijeme mincu 2, zostane X=1, použijeme mincu 1.
- Nefunguje vždy: zoberme mince hodnôt 1,3,4. Pre X=6 najlepšie riešenie je 2 mince: 3+3, ale náš postup (algoritmus) nájde 3 mince 4+1+1
- Ukážeme si algoritmus na základe dyn. programovania, ktorý pre každý vstup nájde najlepšie riešenie
- Zrátame najlepší počet mincí nielen pre X, ale pre všetky možné cieľové sumy 1,2,3,...,X-1,X
- To sa zdá byť ťažšia úloha, ale ukáže sa, že z riešenia pre menšie sumy vieme zostaviť riešenie pre väčšie sumy, takže nám to vlastne pomôže
- Spravíme si tabuľku, kde si pre každú sumu i=0,1,2,...X pamätáme A[i]=najmenší počet mincí, ktoré treba na vyplatenie sumy i
- Ukážme si to na príklade s mincami 1,3,4
i 0 1 2 3 4 5 6 7 8 9 A[i] 0 1 2 1 1 2 2 2 2 3
- Nevypĺňali sme ju žiadnym konkrétnym postupom, nejde o algoritmus
- Ale predstavme si, ze teraz chceme vyplniť A[10].
- V najlepšom riešení je prvá minca, ktorú použijeme 1,3, alebo 4
- ak je prvá minca 1, máme ešte zaplatiť sumu 10-1=9, tú podľa tabuľky vieme najlepšie zaplatiť na 3 mince, takže potrebujeme 4 mince na zaplatenie 10
- ak je prvá minca 3, máme ešte zaplatiť 10-3 = 7, na čo potrebujeme podla tabuľky 2 mince, takže spolu 3 mince na zaplatenie 10
- ak je prvá minca 4, máme ešte zaplatiť 10-4 = 6, na čo treba 2 mince, t.j. 3 mince na 10
- Nevieme, ktorá z týchto možností je naozaj v najlepšom riešení, ale pre druhé dva prípady dostávame menej mincí, takže výsledok budu 3 mince (napr. 3+3+4)
- Zovšeobecníme: A[i] = min { A[i-1]+1, A[i-3]+1, A[i-4]+1 }
- A[11] = min { 3+1, 2+1, 2+1} = min {4, 3, 3 } = 3
- Pre sústavu mincí 1,2,5, máme A[i] = 1+ min { A[i-1], A[i-2], A[i-5] }
- Vo všeobecnosti A[i] = 1+ min { A[i-m_1], A[i-m_2], ..., A[i-m_k] }
- Vzorec treba modifikovať pre malé hodnoty i, ktoré sú menšie ako najväčšia minca, lebo A[-1] a pod. nie je definované
- Zapíšme algoritmus pre všeobecné mince
A[0] = 0;
pre kazde i od 1 po X
min = nekonecno
pre kazde j od 1 po k
ak i >= m_j a A[i-m_j] < min
min = A[i-m_j]
A[i] = 1 + min
vypis A[X]
- Ako nájsť, ktore mince pouzit?
- Pridame druhu tabulku B, kde v B[i] si pamatame, ktora bola najlepsia prva minca, ked sme pocitali A[i] (ak je viac možnosti, zoberieme lubovolnu, napr. najvacsiu)
i 0 1 2 3 4 5 6 7 8 9 10 A[i] 0 1 2 1 1 2 2 2 2 3 3 B[i] - 1 1 3 4 4 3 4 4 4 4
- Potom ak chceme najst napr. mince pre 10, vidime, ze prva bola B[10]=4. Zvysok je 6 a prva minca na vyplatenie 6 je B[6]=3. Zostava nam 3 a B[3]=3. Potom nam uz zostava 0, takze sme hotovi. Takze najlepsie vyplatenie je 4+3+3
- Algoritmus:
Kym X>0 vypis B[X]; X = X-B[X];
- Dynamicke programovanie vo vseobecnosti
- Okrem riesenia celeho problemu, vyriesime aj spustu mensich podproblemov
- Riesenia podproblemov ukladame do tabulky
- Pri rieseni vacsieho podproblemu pouzivame uz vypocitane hodnoty pre mensie podproblemy
- Aka je casova zlozitost?
- Dva parametre: X a k.
- Tabulka velkosti O(X), kazde policko cas O(k). Celkovo O(Xk).
Úvod do pravdepodobnosti
- Myšlienkový experiment, v ktorom vystupuje náhoda, napr. hod ideálnou kockou/korunou
- Výsledkom experimentu je nejaká hodnota (napr. číslo, alebo aj niekoľko čísel, reťazec)
- Túto neznámu hodnotu budeme volať náhodná premenná
- Zaujíma nás pravdepodobnosť, s akou náhodná premenná nadobúda jednotlivé možné hodnoty
- T.j. ak experiment opakujeme veľa krát, ako často uvidíme nejaký výsledok
Príklad 1: hodíme idealizovanou kockou, premenná X bude hodnota, ktorú dostaneme
- Možné hodnoty 1,2,..,6, každá rovnako pravdepodobná
- Píšeme napr. Pr(X=2)=1/6
Príklad 2: hodíme 2x kockou, náhodná premenná X bude súčet hodnôt, ktoré dostaneme
- Možné hodnoty: 2,3,...,12
- Každá dvojica hodnôt (1,1), (1,2),...,(6,6) na kocke rovnako pravdepodobná, t.j. pravdepodobnosť 1/36
- Súčet 5 môžeme dostať 1+4,2+3,3+2,4+1 - t.j. P(X=5) = 4/36
- Súčet 11 môžeme dostať 5+6 alebo 6+5, t.j. P(X=11) = 2/36
- Rozdelenie pravdepodobnosti: (tabuľka udávajúca pravdepodobnosť pre každú možnú hodnotu)
hodnota i: 2 3 4 5 6 7 8 9 10 11 12 Pr(X=i): 1/36 2/36 3/36 4/36 5/36 6/36 5/36 4/36 3/36 2/36 1/36
- Overte, ze súčet pravdepodobností je 1
Stredná hodnota E(X):
- priemer z možných hodnôt váhovaných ich pravdepodobnosťami
- v našom príklade
- Ak by sme experiment opakovali veľa krát a zrátali priemer hodnôt X, ktoré nám vyšli, dostali by sme číslo blízke E(X)
- Iný výpočet strednej hodnoty:
- X=X1+X2, kde X1 je hodnota na prvej kocke a X2 je hodnota na druhej kocke
- , podobne aj E(X2) = 3.5
- Platí, že E(X1+X2)=E(X1) + E(X2) a teda E(X) = 3.5 + 3.5 = 7
- Pozor, pre súčin a iné funkcie takéto vzťahy platiť nemusia, napr. nie je vždy
Pravdepodobnostný model náhodnej sekvencie
- Napríklad chceme modelovať náhodnú DNA sekvenciu dĺžky n s obsahom GC 40%
- Máme vrece s guľôčkami označenými A,C,G,T, pričom guľôčok označených A je 30%, C 20%, G 20% a T 30%.
- Vytiahneme guľôčku, zapíšeme si písmeno, hodíme ju naspäť, zamiešame a opakujeme s ďalším písmenom atď, až kým nevygenerujeme n písmen
- Vytiahnime z mechu 2x guľôčku. Prvé písmeno, ktoré nám vyjde, označme X1 a druhé X2
- Pr(X1=A) = 0.3, Pr(X2=C)=0.2
- Pr(X1=A a X2=C) = Pr(X1=A)*Pr(X2=C) = 0.3*0.2 = 0.06
- T.j. šanca, že dostaneme sekvenciu AC po dvoch ťahoch je 6%
- Ak rátame pravdepodobnosť, že sa dve nezávislé udalosti stanú, ich pravdepodobnosti násobíme. V tomto prípade to, či X1=A je nezávislé od toho, či X2=C
- Pr(X1 je A alebo C) = Pr(X1=A)+Pr(X1=C) = 0.3+0.2 = 0.5
- Pravdepodobnosť, že prvé písmeno bude A alebo C je 50%
- Pravdepodobnosti navzájom sa vylučujúcich udalostí (X1=A a X1=C) sa môžu sčítať, čím dostaneme pravdepodobnosť, že aspoň jedna z nich nastane
- Pr(v sekvencii je aspoň jedno A) = Pr(X1=A alebo X2=A) nemôžeme počítať ako Pr(X1=A)+Pr(X2=A), lebo sa navzájom nevylučujú a prípad, že X1=A a X2=A by sme započítali dvakrát
- Správne je Pr(X1 je A alebo X2 je A) = Pr(X1=A) + Pr(X1 <> A a X2=A) = Pr(X1=A) + Pr(X1 <> A) * Pr(X2=A) = 0.3+0.7*0.3 = 0.51
- Pr(X1=X2) = Pr(X1=X2=A) + Pr(X1=X2=C) + Pr(X1=X2=G) + Pr(X1=X2=T) = 0.3*0.3+0.2*0.2+0.2*0.2+0.3*0.3 = 0.26.
- Ak u označíme pravdepodobnosť u = Pr(X1=A)=Pr(X1=T)=Pr(X2=A)=Pr(X2=T) a v=Pr(X1=C)=Pr(X1=G)=Pr(X2=C)=Pr(X2=G), aký bude vzorec pre Pr(X1=X2)?
Príklad použitia modelu: Máme krátky primer AACAT. Koľko bude mať v priemere výskytov v sekvencii dĺžky 1000 v našom modeli?
- Pravdepodobnosť, ze AACAT je v náhodnej sekvencii hneď na začiatku je Pr(X1=A a X2=A a X3=C a X4=A a X5=A) = 0.3*0.3*0.2*0.3*0.3 = 0.00162
- Rovnaká pravdepodobnosť aj na pozícii 2,3,...96
- Nech V je počet výskytov v celej sekvencii (náhodná premenná s možnými hodnotami 0,1,...,996, aj keď napr. 996 to určite nemôže byť)
- Ideálne by sme chceli spočítať celú tabuľku pravdepodobností pre V, ale uspokojíme sa aj so strednou hodnotou E(V)
- Nech Vi je počet výskytov na pozícii i (co je vzdy 0 alebo 1)
-
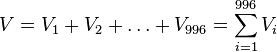
-
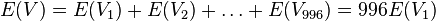
-
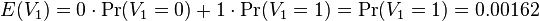
- E(V) = 96*0.00162 = 1.61352
- Takze primer AACAT sa v priemere bude v nahodnej sekvencii dlzky 1000 s 40% obsahov GC vyskytovat v priemere cca 1,6 krat
- Primery byvaju dlhsie, takze sanca nahodnych vyskytov je ovela mensia, co je to co vacsinou chceme (chceme primer cielit na konkretnu poziciu, nie na vela nahodnych zhod)
Použitie pravdepodobnosti na analýzu potrebného pokrytia pri sekvenovaní
Nerobili sme, uvedené pre zaujímavosť, pozri cvičenia pre informatikov
CI03
Dynamické programovanie
- Pozri Cvičenia pre biológov
Uvod do proteomiky
- Viac informacii: [5], Bafna, Reinert 2004
- Pozri tiež prezentáciu k cvičeniu
Gélová elektroforéza (gel electrophoresis)
- Izolovanie jednotlivých proteínov, porovnávanie ich množstva.
- Negatívne nabité proteíny migrujú v géli v elektrickom poli. Väčšie proteíny migrujú pomalšie, dochádza v oddeleniu do pruhov. Táto metóda sa používa aj na DNA a RNA. Pre proteíny možno tiež robiť 2D gél (podľa hmotnosti a náboja).
- Bioinformatický problém: zisti, ktoré fliačiky na dvoch 2D géloch zodpovedajú tým istým proteínom.
- Automatizovanejšia technológia: kvapalinová chromatografia (liquid chromatography) - separácia proteínov v tenkom stĺpci
Hmotnostná spektrometria (mass spectrometry)
- Hmotnostná spektrometria meria pomer hmostnosť/náboj molekúl vo vzorke.
- Používa sa na identifikáciu proteínov, napr. z 2D gélu.
- Proteín nasekáme enzýmom trypsín (seká na [KR]{P}) na peptidy
- Meriame hmostnosť kúskov, porovnáme s databázou proteínov.
- Tandemová hmotnostná spektrometria (MS/MS) ďalej fragmentuje každý kúsok a dosiahne podrobnejšie spektrum, ktoré obsahuje viac informácie
- v niektorých prípadoch vieme sekvenciu proteínu určiť priamo z MS/MS, bez databázy proteínov
Sekvenovanie proteinov pomocou MS/MS
Vsetky hmotnosti budeme povazovat za cele cisla
Vstup:
- celková hmotnosť peptidu M,
- hmotnosti aminokyselín a[1],...,a[20],
- spektrum ako tabuľka f[0],...,f[M], ktorá hmotnosti m určí skóre f[m] podľa signálu v okolí príslušného bodu grafu
Označenie:
- Uvažujme postupnosť aminokyselín
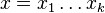
- Nech
![m(x)=\sum _{{j=1}}^{k}a[x_{j}]](/vyuka/mbi/images/math/5/1/e/51e47d7db5cc3125eee0a57b9e19c12c.png) je hmotnosť x
je hmotnosť x
- Nech
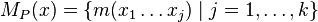 sú hmotnosti prefixov x
sú hmotnosti prefixov x
- Nech
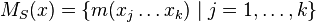 sú hmotnosti sufixov x
sú hmotnosti sufixov x
Problém 1
Berme do uvahy len b-iony, ktore zodpovedaju hmotnosti prefixu
Výstup:
- postupnosť aminokyselín x taká, že
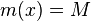 a
a ![\sum _{{m\in M_{P}(x)}}f[m]](/vyuka/mbi/images/math/e/f/c/efca77d5389ed7769b65877ff30a2c69.png) je maximálna možná
je maximálna možná
- Chceme teda najst peptid, ktory maximalizuje sucet skore svojich prefixov
Riešenie
- Dynamicke programovanie s podproblemom S[m] je skore najlepsieho prefixu s hmotnostou m
- Rekurencia? Zlozitost? Je to polynomialny algoritmus? (Aky velky je vlastne vstup?)
Problém 2
Berme do uvahy aj y-iony, ktore meraju hmotnost sufixu, scitame skore prefixov a sufixov
Výstup:
- postupnosť aminokyselín x taká, že a
![\sum _{{m\in M_{P}(x)}}f[m]+\sum _{{m\in M_{S}(x)}}f[m]](/vyuka/mbi/images/math/6/9/9/699ef640d8249b3456c1241580179964.png) je maximálna možná
je maximálna možná
Riešenie
- pouzijeme upravenu skorovaciu tabulku g[m]=f[m]+f[M-m] a algoritmus pre problem 1
Problem tejto formulacie:
- jeden signal sa moze ratat dvakrat, raz ako b-ion, raz ako y-ion, algoritmus ma tendenciu pridavat taketo artefakty
Problém 3
Ak hmotnost nejakeho prefixu a nejakeho sufixu su rovnake, zarataj ich skore iba raz (skore peptidu je skore mnoziny hmotnosti jeho prefixov a sufixov)
Výstup:
- postupnosť aminokyselín x taká, že a
![\sum _{{m\in M_{P}(x)\cup M_{S}(x)}}f[m]](/vyuka/mbi/images/math/6/6/5/665a026ed145fe16ec6b27b6044972b8.png) je maximálna možná
je maximálna možná
Riesenie:
- Ina formulacia: maximalizujeme
![\sum _{{m\in M_{p}(x)\cup M_{S}(x),m\leq M/2}}h[m]](/vyuka/mbi/images/math/d/2/0/d20e9653e2a74d027573395f12f2f0f1.png)
-
![h[m]=\left\{{\begin{array}{ll}f[m]+f[M-m]&{\mbox{ak }}m<M/2\\f[m]&{\mbox{ak }}m=M/2\end{array}}\right.](/vyuka/mbi/images/math/6/b/9/6b9baa42bbaae35399de43cab3712d19.png)
- Definuj novy podproblem: S[p,s] je najlepsie skore, ktore moze dosiahnut prefix s hmotnostou p a sufix s hmotnostou s, kde 0<=p,s<=M/2,
- Rekurencia
![S[p,s]=\left\{{\begin{array}{ll}\max _{{i=1\dots 20}}S[p,s-a[i]]+h[s]&{\mbox{ak }}p<s\\\max _{{i=1\dots 20}}S[p-a[i],s]+h[p]&{\mbox{ak }}p>s\\\max _{{i=1\dots 20}}S[p-a[i],s]&{\mbox{ak }}p=s\\\end{array}}\right.](/vyuka/mbi/images/math/f/b/1/fb198e39be6b286cd92f4e0a8c0bbddf.png)
- Ako ukoncime dynamicke programovanie? Zlozitost?
- Zrychlenie: staci uvazovat s od p-w po p+w kde w je maximalna hmotnost aminokyseliny
Detekcia znamych proteinov pomocou MS (nerobili sme)
- Predikcia spektra pre dany peptid, porovnanie s realnym spektrom, zlozite skorovacie schemy
- Filtrovanie kandidatov na proteiny, ktore obsahuju peptidy s pozorovanou hmotnostou
- Problem: mame danu databazu proteinov a cielovu hmotnost peptidu M, pozname hmotnost kazdej aminokyseliny. Najdite vsetky podretazce s hmotnostou M.
- Databazu proteinov si vieme predstavit aj ako postupnost cisel - hmotnosti aminokyselin, hladame intervaly so suctom M.
- Trivialny algoritmus: zacni na kazdej pozicii, pricitavaj kym nedosiahnes hmotnost>=M. Zlozitost? Vieme zlepsit?
- Predspracovanie: pocitajme hmotnosti vsetkych podretazcov, potom vyhladajme binarne. Zlozitost?
- Zlozitejsi alg. s predspracovanim pomocou FFT (Fast Fourier Transform) Bansal, Cieliebak, Liptak 2004
CB03
Zarovnávanie sekvencií, opakovanie
- Uvažujme skórovanie zhoda +2, nezhoda -1, medzera -1
- Reťazce TAACGG a CACACT
Globálne zarovnanie
- Rekurencia: A[i,j] = max {A[i-1,j]-1, A[i,j-1]-1, A[i-1,j-1]+s(x_i, y_j) }, pričom A[0,i]=-i, A[i,0]=-i
C A C A C T
0 -1 -2 -3 -4 -5 -6
T -1
A -2
A -3
C -4
G -5
G -6
Lokálne zarovnanie
- Rekurencia: A[i,j] = max {0, A[i-1,j]-1, A[i,j-1]-1, A[i-1,j-1]+s(x_i, y_j) }, pričom A[0,i]=0, A[i,0]=0
C A C A C T
0 0 0 0 0 0 0
T 0
A 0
A 0
C 0
G 0
G 0
Dotploty
- Dotplot je graf, ktory ma na kazdej osi jednu sekvenciu a ciarky zobrazuju lokalne zarovnania (cesty v matici)
- Niekoľko príkladov dotplotov: pdf
- Prvé príklady dotplotov porovnavaju rozne mitochondrialne genomy
- Tieto boli vytvorene pomocou nastroja YASS http://bioinfo.lifl.fr/yass/yass.php
- Dalsi priklad je zarovnanie genu Oaz Drosophila zinc finger s genomickym usekom chr2R:10,346,241-10,352,965
- Trochu iny dotplot, ktory funguje pre proteiny a nerobi lokalne zarovnania, iba spocita skore bez medzier v kazdom okne danej vysky a nakresli ciaru ak pre kroci urcenu hodnotu
- http://emboss.bioinformatics.nl/cgi-bin/emboss/dotmatcher
- Vyskusame protein escargot voci sebe s hodnotami http://pfam.sanger.ac.uk/protein/ESCA_DROME window 8 threshold 24
- Pomocou YASSu vyskusame kluster zhlukov PRAME z ludskeho genomu
Praktické cvičenie pri počítači: dotploty
Yass a dotploty
- Program Yass hlada lokalne zarovnania v DNA sekvenciach, zobrazuje vo forme dot plotov
- Na stranke UCSC genome browseru http://genome-euro.ucsc.edu/ si zadajte ludsky genom, verziu hg38
- V druhom okne/tabe si otvorte YASS server na adrese http://bioinfo.lifl.fr/yass/yass.php
- V genome browseri zadajte región chr21:9,180,027-9,180,345
- tento región obsahuje Alu repeat. Tieto opakovania tvoria cca 10% ľudského genómu, viac ako milión kópií
- zobrazte si DNA sekvenciu tohto useku takto: na hornej modrej liste zvolte View, potom v podmenu DNA, na dalsej obrazovke tlacidlo get DNA
- DNA sekvenciu Alu opakovania chceme zarovnat samu k sebe programom YASS
- DNA sekvenciu Alu opakovania skopirujte do okienka "Paste your sequences" v stranke Yass-u a dvakrat stlacte tlacidlo Select vedla okienka
- Nizsie v casti "Selected DNA sequence(s)" by sa Vam malo v oboch riadkoch objavit "Pasted file 1"
- Nizsie v casti "Parameters" zvolte "E-value threshold" 0.01 a stlacte "Run YASS"
- Vo vysledkoch si pozrite Dotplot, co z neho viete usudit o podobnosti jednotlivych casti Alu opakovania?
- Vo vysledkoch si pozrite Raw: blast, ake su suradnice opakujucej sa casti a kolko zarovnanie obsahuje zhod/nezhod/medzier? (Pozor, prve zarovnanie je cela sekvencia sama k sebe, druhe je asi to, co chcete)
- V genome browseri zadajte poziciu chr21:8,552,000-8,562,000 (10kb sekvencie na chromozome 21, s niekolkymi vyskytmi Alu)
- Chceme teraz porovnat tento usek genomu so sekvenciou Alu pomocou YASSu
- Ako predtym si stiahnite DNA sekvenciu tohto useku
- V YASSe chodte sipkou spat na formular
- Skopirujte DNA sekvenciu do YASSoveho formulara, do okienka vpravo (vyznacit si ju mozete klavesovou kombinaciou Ctrl-A alebo Select All v menu Edit),
- V casti formulara Selected DNA sequence(s) stlacte Remove pri hornom riadku
- Pri pravom okienku, kam ste nakopirovali sekvenciu, stlacte Select
- Zase stlacte Run YASS
- Pozrite si vysledok ako Dotplot, kolko opakovani Alu ste nasli? Preco je jedno cervene?
- Pozrite si Raw: blast, na kolko percent sa podoba najpodobnejsia a na kolko druha najpodobnejsia kopia?
CI04
Opakovanie dynamického programovania pre globálne zarovnanie
Uvažujme napríklad skórovanie zhoda +1, nezhoda -1, medzera -1 a vstupné sekvencie 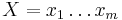 a
a 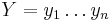 . Nech s(x,y) je skóre písmen x a y, t.j. 1 ak sa zhodujú a -1 ak nie. Máme rekurenciu:
. Nech s(x,y) je skóre písmen x a y, t.j. 1 ak sa zhodujú a -1 ak nie. Máme rekurenciu:
![A[i,j]=\max \left\{A[i-1,j-1]+s(x_{i},y_{j}),A[i-1,j]-1,A[i,j-1]\right\}](/vyuka/mbi/images/math/1/6/f/16f1ddd6bdc5d2a31e14184fdad3cf04.png)
- Ako presne by sme implementovsali?
- Ako spocitame maticu spatnych sipok B?
- Aka je casova a pamatova zlozitost?
Reprezentácia pomocou grafu
Takéto dynamické programovanie vieme reprezentovať vo forme acyklického orientovaného grafu:
- vrchol (i,j) pre každé
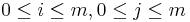 , t.j. pre každé políčko dyn. prog. tabuľky
, t.j. pre každé políčko dyn. prog. tabuľky
- hrana z (i-1,j-1) do (i,j) s cenou
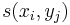
- hrana z (i-1,j) do (i,j) s cenou -1
- hrana z (i,j-1) do (i,j) s cenou -1
- súčet súradníc na každej hrane rastie, graf teda nemôže obsahovať cyklus, je acyklický
- každá cesta z (0,0) do (m,n) zodpovedá zarovnaniu, jej cena je cenou zarovnania (každá hrana jeden stĺpec)
- optimálne zarovnanie teda zodpovedá ceste s maximálnou cenou
Krátka vsuvka o acyklických orientovaných grafoch
- Mame dany acyklicky orientovany graf s ohodnotenymi hranami a startovaci vrchol s, koncovi vrchol t a chceme najst cestu s max. cenou z s do t.
- Hladanie cesty s maximalnou cenou je vo vseobecnosti NP-tazke (podobne na Hamiltonovsku cestu)
- V acyklickom grafe to vsak vieme riesit efektivne
- Najskor si graf zotriedime topologicky, t.j. usporiadame vrcholy tak, aby kazda hrana isla z vrcholu z mensim cislom do vrcholu s vacsim cislom. To sa da modifikaciou prehladavania do hlbky v case O(|V|+|E|)
- Potom pocitame dynamickym programovanim, kde A[u] je dlzka najdlhsej cesty z s do u:
![A[u]=\max _{{v:v\rightarrow u\in E}}A[v]+c(v\rightarrow u)](/vyuka/mbi/images/math/6/d/9/6d90a7118031848ff55c4204bc8b4552.png)
pricom na zaciatku nastavime A[s]=0 a na konci mame cenu cesty v A[t].
- Cas vypoctu je O(|V|+|E|)
- Vsimnime si, ze tiez dostaneme najdlhsie cesty z s do vsetkych vrcholov.
Ak tento algoritmus nasadime na graf pre globalne zarovnanie, dostavame presne nasu rekurenciu (topologicke triedenie mozno vynechat - poradie zhora dole a zlava doprava je topologicky utriedene). Vyhoda je, ze mozeme modifikaciou grafu ziskavat riesenia roznych pribuznych problemov bez toho, aby sme vzdy vymyslali novu rekurenciu.
Lokálne zarovnanie
- Zarovnanie moze zacat a skoncit hocikde v matici
- Pridaj startovaci vrchol s, koncovy vrchol t
- Pridaj hrany s->(i,j) a (i,j)->t s cenou 0 pre kazde (i,j)
- Opat ekvivalentne s rekurenciou z prednasky
Variant: chceme zarovnat cely retazec X k nejake casti retazca Y (napr. mapovanie sekvenovacich readov na genom)
- Iba zmenime hrany z s a hrany do t (ako?)
Afínne skóre medzier
- Napr. otvorenie medzery o=-3, pokracovanie medzery e=-1
A - - - T C G A C G C T C C 1 -3 -1 -1 1 1 -1
Nesprávne riešenie pomocou dynamického programovania
Pouzijeme bezne dynamicke programovanie pre globalne zarovnanie, ale v rekurencii zmenime vypocet penalty za medzeru:
![A[i,j]=\max \left\{A[i-1,j-1]+s(x_{i},y_{j}),A[i-1,j]+c(i-1,j,hore),A[i,j-1]+c(i,j-1,vlavo)\right\}](/vyuka/mbi/images/math/8/c/c/8cc47a0f26c209544027bcd2bbe77818.png)
- c(i,j,s) = o, ak v policku A[i,j] mame sipku s
- c(i,j,s) = e, ak v policku A[i,j] mame inu sipku
Preco toto riesenie nefunguje?
- Co ak pre policko (i,j) je viac rovnako dobrych rieseni s roznymi sipkami?
- Co ak pre policko (i,j) je najlepsie riesenie so sipkou napr. sikmo, ale druhe najlepsie je len 1 horsie a ma sipku hore?
Toto je obvykla chyba pri dynamickom programovani:
- aby bolo dynamicke programovanie spravne, musi platit, ze optimalne riesenie vacsieho podproblemu musi obsahovat optimalne riesenie mensieho podproblemu
Správne riešenie pomocou dynamického programovananie
Riesenie 1:
- Pridame hrany pre cele suvisle useky medzier so spravnou cenou
- (i,j)->(i,k) s cenou o+(k-j)e
- (i,j)->(k,j) s cenou o+(k-i)e
- Cas O(mn(m+n)), t.j. kubicky
- pozor, mame aj cesty, ktore nezopodvedaju ziadnemu spravnemu skore, napr. (i.j)->(i+1,j)->(i+2,j) ma cenou 2o, ale ma mat o+e. Nastastie hrana (i,j)->(i+2,j) ma vyssiu cenu, takze ta dlhsia cesta sa nepouzije.
Riesenie 2:
- ztrojnasobime kazdy vrchol
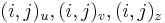
- v indexe si pamatame, odkial sme do (i,j) prisli (u=uhlopriecne, v=vodorovne, z=zvislo)
- ak ideme napr. z
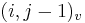 do
do 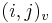 , pokracujeme v uz existujucej medzere, takze skore je e
, pokracujeme v uz existujucej medzere, takze skore je e
- ak ideme napr. z
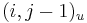 do , zaciname novu medzeru, takze skore je o
do , zaciname novu medzeru, takze skore je o
- ake vsetky hrany teda mozeme mat? Kolko je spolu v grafe hran a vrcholov a aka je zlozitost algoritmu?
Lineárna pamäť: Hirshbergov algoritmus 1975
- Klasicke dynamicke programovanie potrebuje cas O(nm)
- Trivialna implementacia tiez pouzije pamat O(mn) - uklada si celu maticu A, pripadne maticu B so sipkami naspat
- Na vypocet matice A nam z stacia dva riadky tejto matice: riadok i pocitam len pomocou riadku i-1, starsie viem zahodit
- Ale ak chcem aj vy[isat zarovnanie, stale potrebujem pamat O(mn) na maticu sipok B
- Hirschbergov algoritmus znizi pamat na O(m+n), zhruba zdvojnasobi cas (stale O(mn))
- Prejdeme celú maticu a spočítame maticu A. Zapamätáme si, kde moja cesta prejde cez stredný riadok matice
- Nech B_k[i,j] je najväčší index v riadku k, cez ktorý prechádza najkratšia cesta z (0,0) do (i,j)
- Ako vieme B_k[i,j] spočítať?
- ak A[i,j] = A[i-1,j-1]+w(S[i],T[j])$, potom B_k[i,j]=B_k[i-1,j-1].
- ak A[i,j]=A[i-1,j]+1, potom B_k[i,j]=B_k[i-1,j].
- ak A[i,j]=A[i,j-1]+1, potom B_k[i,j]=B_k[i,j-1]
- Toto platí, ak i > k. Pre i=k nastavíme B_k[i,j]=j
- Ak už poznáme A[i-1,*] a B_k[i-1,*], vieme spočítať A[i,*] a B_k[i,*].
- Stacia nam teda iba dva riadky matice A a B_k
- Nech k'=B_k[m,n]. Potom v optimálnom zarovnaní sa S[1..k] zarovná s T[1..k'] a S[k+1..m] s T[k'+1..n].
- Toto použijeme na rekurzívny algoritmus na výpočet zarovnania:
optA(l1, r1, l2, r2) { // align S[l1..r1] and T[l2..r2]
if(r1-l1 <= 1 || r2-l2 <=1)
solve using dynamic programming
else {
k=(r-l+1)/2;
for (i=0; i<=k; i++)
compute A[i,*] from A[i-1,*]
for (i=k+1; i<=r-l+1; i++)
compute A[i,*], B_k[i,*] from A[i-1,*], B_k[i-1,*]
k2=B_k[r1-l1-1,r2-l2-1];
optA(l1, l1+k-1, l2, l2+k2-1);
optA(l1+k, r2, l2+k2, r2);
}
}
Casova zlozitost:
- Označme si N=nm (súčin dĺžky dvoch daných reťazcov).
- Na hornej úrovni rekurzie spúšťame dynamické programovanie pre celú maticu -- čas bude $cN$.
- Na druhej urovni mame dva podproblemy, velkosti N1 a N2, pricom N1+N2<=0.5*N (z kazdeho stlpca matice A najviac polovica riadkov pocitana znova)
- Na tretej urovni mame 4 podproblemy N11, N12, N21, N22, pricom N11+N12 <= 0.5*N1 a N21+N22 <= 0.5*N2 a teda celkovy sucet podproblemov na druhej urvni je najviac N/4.
Na stvrtej urovni je sucet podproblemov najviac N/8 atd, Dostavame geometricky rad cN+cn/2+cN/4+... ktoreho sucet je 2cN
Vypísanie všetkých najlepších riešení
- Namiesto jednej spatnej sipky si pamatame vsetky, ktore v danom A[i,j] viedli k maximalnej cene
- Potom mozeme rekurzivne prehladavat a vypisovat vsetky cesty z (m,n) do (0,0) ktore pozostavaju iba zo zapamatanych hran
- Cas na vypisanie jednej cesty je polynomialny, ale ciest moze byt exponencialne vela!
- Mozno namiesto toho chceme len pocet takych ciest, alebo vsetky dvojice pismen, ktore mozu byt spolu zarovnane v niektorom optimalnom zarovnani
CB04
Príklady praktických programov
Pozrime sa na niekolko nastrojov, vsimnime si, ake poskytuju nastavenia a co vypisuju na vystupe, dajme to do suvisu s prednaskami
- viacnasobne zarovnania o tyzden
Plné dynamické programovanie
- Balicek emboss, obsahuje programy na klasicke dynamicke programovanie (needle - globalne, water - lokalne), najdu sa na stranke EBI http://www.ebi.ac.uk/Tools/psa/
- porovnanie lokalneho a globalneho zarovnania
- Dva proteiny s kinase doménou zarovnáme lokálne, globálne a globálne s tým, že neplatíme za medzery na koncoch
>sp|P50520|VPS34_SCHPO Phosphatidylinositol 3-kinase vps34 OS=Schizosaccharomyces pombe (strain 972 / ATCC 24843) GN=vps34 PE=2 SV=2 >tr|B1AKP8|B1AKP8_HUMAN FK506 binding protein 12-rapamycin associated protein 1 OS=Homo sapiens GN=FRAP1 PE=4 SV=1
- sekvencie a vysledne zarovnania: CB-aln-dp
- vo vysledku si vsimnime, kolko ma kazde z nich %identity, %gaps, a kam sa zarovna sekvencia na pozicii 53 v spodnej sekvencii (NSESEAE) a kam sekvencia na pozicii 395 (EDLRQDE)
Lokalne zarovnanie
Length: 645
Identity: 124/645 (19.2%)
Similarity: 221/645 (34.3%)
Gaps: 211/645 (32.7%)
Score: 226.0
VPS34_SCHPO 235-738
B1AKP8_HUMAN 53-627
235 NLDSPAE
|.:|.||
53 NSESEAE
549 DDLRQDQ
:|||||:
395 EDLRQDE
Globalne zarovnanie
Length: 948
Identity: 167/948 (17.6%)
Similarity: 292/948 (30.8%)
Gaps: 341/948 (36.0%)
Score: 130.5
VPS34_SCHPO 1-801
B1AKP8_HUMAN 1-754
102 NDEEVYE
|.|...|
53 NSESEAE
549 DDLRQDQ
:|||||:
395 EDLRQDE
Globalne zarovnanie s nulovou penaltou za medzeru na koncoch
Length: 1060
Identity: 138/1060 (13.0%)
Similarity: 245/1060 (23.1%)
Gaps: 565/1060 (53.3%)
Score: 206.0
VPS34_SCHPO 234-801
B1AKP8_HUMAN 1-674
265 KIRKELESIL
....|.||..
53 NSESEAESTE
549 DDLRQDQ
:|||||:
395 EDLRQDE
NCBI Blast
- NCBI BLAST http://blast.ncbi.nlm.nih.gov/ vela roznych nastrojov (porovnavanie DNA vs proteiny, pripadne translacia DNA na protein v 6 ramcoch)
- Heuristicky algoritmus, moze niektore zarovnania vynechat
- rozne nastavenia, vystup E-value
Low complexity masking: nepouzivat pri hladani jadier zarovnania regiony v ktorych sa velakrat opakuje ta ista aminokyselina
- Priklad (z ucebnice Zvelebil and Baum):
>sp|P04156|PRIO_HUMAN Major prion protein OS=Homo sapiens GN=PRNP PE=1 SV=1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV ILLISFLIFLIVG
Bez maskovania vypise najpr aj toto zarovnanie:
>ref|NP_065842.1| serine/threonine-protein kinase TAO1 isoform 1 [Homo sapiens]
Length=1001
Score = 45.1 bits (105), Expect = 1e-06, Method: Composition-based stats.
Identities = 26/61 (43%), Positives = 27/61 (44%), Gaps = 11/61 (18%)
Query 38 YPGQGSPGGNRYPPQGGGG--WGQPHGG---GWGQPHGGG---WGQPHGGGWGQPHGGGWG 90
YPG G + P GG G WG P GG WG P GG WG P G G P G G
Sbjct 904 YPGAS---GWSHNPTGGPGPHWGHPMGGPPQAWGHPMQGGPQPWGHPSGPMQGVPRGSSMG 961
Score = 40.0 bits (92), Expect = 4e-05, Method: Composition-based stats.
Identities = 25/62 (40%), Positives = 25/62 (40%), Gaps = 10/62 (16%)
Query 26 PKPGGW--NTGGSRYPGQGSPGGNRYPPQGGGGWGQPHGGG---WGQPHGGGWGQPHGGGWG 82
P GW N G P G P G PPQ WG P GG WG P G G P G
Sbjct 905 PGASGWSHNPTGGPGPHWGHPMGG--PPQA---WGHPMQGGPQPWGHPSGPMQGVPRGSSMG 961
Ak zapneme maskovanie, toto zarovnanie uz nenajde, v zarovnani sameho so sebou sa objavia male pismena alebo Xka:
>ref|NP_000302.1|major prion protein preproprotein [Homo sapiens]
Length=253
Score = 520 bits (1340), Expect = 0.0, Method: Compositional matrix adjust.
Identities = 253/253 (100%), Positives = 253/253 (100%), Gaps = 0/253 (0%)
Query 1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYppqggggwgqp 60
MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP
Sbjct 1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP 60
Query 61 hgggwgqphgggwgqphgggwgqphgggwgqgggTHSQWNKPSKPKTNMKHMagaaaaga 120
HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA
Sbjct 61 HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA 120
Query 121 vvgglggymlgsamsRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV 180
VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV
Sbjct 121 VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV 180
Query 181 NITIKQHtvttttkgenftetDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSsppv 240
NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV
Sbjct 181 NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV 240
Query 241 illisfliflivG 253
ILLISFLIFLIVG
Sbjct 241 ILLISFLIFLIVG 253
BLAT, chains, nets v UCSC browseri
- Program BLAT v UCSC browseri http://genome.ucsc.edu/ rychlo vyhladava sekvencie v genome, ale nevie najst slabsie podobnosti
- Vhodne pouzitie: zarovnanie EST ku genomu, presne urcenie suradnic nejakej sekvencie, a pod.
- Net tracky v UCSC genome browseri nam umoznuju prechadzat medzi homologickymi oblastami roznych genomov
Praktické cvičenie pri počítači: BLAT vs BLAST
BLAT/BLAST
- Sekvencia uvedena nizsie vznikla pomocou RT-PCR na ľudských cDNA knižniciach
- Choďte na UCSC genome browser http://genome.ucsc.edu/ , na modrej lište zvoľte BLAT, zadajte túto sekvenciu a hľadajte ju v ľudskom genóme. Akú podobnosť (IDENTITY) má najsilnejší nájdený výskyt? Aký dlhý úsek genómu zasahuje? (SPAN). Všimnite si, že ostatné výskyty sú oveľa kratšie.
- V stĺpci ACTIONS si pomocou Details môžete pozrieť detaily zarovnania a pomocou Browser si pozrieť príslušný úsek genómu.
- V tomto úseku genómu si zapnite track Vertebrate net na full a kliknutím na farebnú čiaru na obrázku pre tento track zistite, na ktorom chromozóme sliepky sa vyskytuje homologický úsek.
- Skusme tu istu sekvenciu zarovnat ku genomu sliepky programom Blat: stlacte najprv na hornej modrej liste Genomes, zvolte Vertebrates a Chicken a potom na hornej liste BLAT. Do okienka zadajte tu istu sekvenciu. Akú podobnosť a dĺžku má najsilnejší nájdený výskyt teraz? Na ktorom je chromozóme?
- Skúsme to isté v NCBI blaste: Choďte na http://blast.ncbi.nlm.nih.gov/ zvoľte nucleotide blast, database others a z menu reference genomic sequence, organism chicken (taxid:9031), program blastn
- Aka je dlzka, identity a E-value najlepsieho zarovnania? Na ktorom je chromozome?
RT PCR sekvencia z cvičenia vyššie
AACCATGGGTATATACGACTCACTATAGGGGGATATCAGCTGGGATGGCAAATAATGATTTTATTTTGAC TGATAGTGACCTGTTCGTTGCAACAAATTGATAAGCAATGCTTTCTTATAATGCCAACTTTGTACAAGAA AGTTGGGCAGGTGTGTTTTTTGTCCTTCAGGTAGCCGAAGAGCATCTCCAGGCCCCCCTCCACCAGCTCC GGCAGAGGCTTGGATAAAGGGTTGTGGGAAATGTGGAGCCCTTTGTCCATGGGATTCCAGGCGATCCTCA CCAGTCTACACAGCAGGTGGAGTTCGCTCGGGAGGGTCTGGATGTCATTGTTGTTGAGGTTCAGCAGCTC CAGGCTGGTGACCAGGCAAAGCGACCTCGGGAAGGAGTGGATGTTGTTGCCCTCTGCGATGAAGATCTGC AGGCTGGCCAGGTGCTGGATGCTCTCAGCGATGTTTTCCAGGCGATTCGAGCCCACGTGCAAGAAAATCA GTTCCTTCAGGGAGAACACACACATGGGGATGTGCGCGAAGAAGTTGTTGCTGAGGTTTAGCTTCCTCAG TCTAGAGAGGTCGGCGAAGCATGCAGGGAGCTGGGACAGGCAGTTGTGCGACAAGCTCAGGACCTCCAGC TTTCGGCACAAGCTCAGCTCGGCCGGCACCTCTGTCAGGCAGTTCATGTTGACAAACAGGACCTTGAGGC ACTGTAGGAGGCTCACTTCTCTGGGCAGGCTCTTCAGGCGGTTCCCGCACAAGTTCAGGACCACGATCCG GGTCAGTTTCCCCACCTCGGGGAGGGAGAACCCCGGAGCTGGTTGTGAGACAAATTGAGTTTCTGGACCC CCGAAAAGCCCCCACAAAAAGCCG
Dynamické programovanie v Exceli
Práca so vzorcami v tabuľkovom procesore (Excel, LibreOffice, ...)
- Okrem konkrétnych hodnôt, napr. 0.3, môžu byť aj vzorce, ktoré začínajú =, napr =0.3*0.3 dá do políčka 0.09 (* znamená násobenie)
- Vo vzorcoch môžeme používať aj hodnoty z iných políčok, napr. =A2+B2 dáme do políčka C2, zobrazí sa tam súčet
- Ak políčko so vzorcom skopírujeme do iného políčka, Excel sa snaží uhádnuť, ako zmeniť vzorec
- Ak sme v C2 mali =A2+B2 a skopírovali sme to do C3, vzorec sa zmení na =A3+B3
- Ak niektoré adresy políčok majú zostávať rovnaké aj pri kopírovaní, dáme pred písmeno aj číslo $,
- Ak v C2 máme =A2+$B$2 a skopírujeme to do C3, dostaneme =A3+B2
- Dolár môžeme dať aj pred iba jednu súradnicu (stĺpec alebo riadok), tá sa potom nebude pri kopírovaní meniť
Späť k minciam
- nerobili sme, uvedené pre zaujímavosť
- Vráťme sa k príkladu s rozmieňaním mincí a skúsme si ho "naprogramovať" v Exceli, resp. spreadsheet aplikácii v OpenOffice
- Vseobecna formulacia:
- Vstup: hodnoty k minci m_1,m_2,...,m_k a cielova suma X (vsetko kladne cele cisla)
- Vystup: najmensi pocet minci, ktore potrebujeme na zaplatenie X
- My pouzijeme mince hodnot 1,3,4
- Spravime si tabulku, kde si pre kazdu sumu i=0,1,2,...X pamatame A[i]=najmensi pocet minci, ktore treba na vyplatenie sumy i (ak je viac moznosti, zoberieme lubovolnu, napr. najvacsiu)
i 0 1 2 3 4 5 6 7 8 9 A[i] 0 1 2 1 1 2 2 2 2 3
- vzorec A[i] = min { A[i-1]+1, A[i-3]+1, A[i-4]+1 }
- aby sme nemuseli zvlast uvazovat hodnoty mensie ako 4, (kde sa neda A[i-4]), urcime si A[-1], A[-2] atd ako nejake velke cislo (napr 100), takze vzorec plati pre vsetky i>0
i -4 -3 -2 -1 0 1 2 3 4 5 6 7 8 9 A[i] 100 100 100 100 0 1 2 1 1 2 2 2 2 3
- v exceli si najskor spravime horny riadok tabulky
- do nejakeho policka (napr, B4) zapiseme prvu hodnotu (-4)
- do susedneho C4 zapiseme vzorec =B4+1, dostaneme hodnotu -3
- vzorce zacinaju znamienkom =
- B4 je suradnica policka o jedno vlavo, k nej pripocitame 1
- policko C4 nakopirujeme do riadku kolkokrat chceme, dostaneme hodnoty -2, -1, 0, 1,...
- kopirovat sa da tahanim laveho dolneho rohu okienka
- vzorec sa automaticky posuva na =C4+1, =D4+1, atd
- o riadok nizsie do B5..E5 napiseme hodnotu 100 (okienka A[-4]..A[-1])
- do F5 dame 0 (okienko A[0] nasej tabulky)
- do G5 napiseme vzorec =MIN(F5+1,D5+1,C5+1), t.j. A[1] = min(A[1-1]+1,A[1-3]+1,A[1-4]+1)
- tento vzorec potom nakopirujeme do riadku tabulky
- F5 sa bude posuvat na G5, H5,... a podobne ostatne dva cleny
Cvičenie:
- Ako by sme zmenili na inu mincovu sustavu, napr. 1,2,5?
- Stiahnite si subor zo stranky predmetu a skuste si tuto zmenu urobit [6]
Zarovnávanie sekvencií v Exceli
- skusme si dynamicke programovanie pre globalne zarovnanie naprogramovat v Exceli
- budeme postupovat podobne ako pri minciach, ale potrebujeme dve specialne funkcie: MID(text,od,dlzka) z textu vyberie urcitu cast. Pomocou toho si vstupny text rozdelime na jednotlive pismena, ktore si napiseme do zahlavia tabulky
- vsimnite si pouzivanie dolarov v nazvoch policok: ak je pred menom stlpca alebo riadku $, tento sa neposuva ked vzorec kopirujem do inych policok
- IF(podmienka,hodnota1,hodnota2) vyberie bud hodnotu 1 ak je podmienka splnena alebo hodnotu2 ak nie je. Napr IF(F$8=$B12 ,1,-1) zvoli skore +1 ak sa hodnota v F8 rovna hodnote v B12 a skore -1 ak sa nerovnaju.
Cvicenie:
- Zmente tabulku tak, aby skore pre zhody, nezhody a medzery bolo dane bunkami B1, B2 a B3 tabulky. Staci zmenit vzorce a policka D9, C10 a D10 a nakopirovat do zvysku tabulky. Ake bude skore najlepsieho zarovnania sekvencii AACGTA a ACACCTA ak skore nezhody je -2 a medzery -3?
- Ako treba zmenit vzorce, aby sme pocitali lokalne zarovnanie?
- Subor najdete tu
Skórovacie matice
Chceme urcit skorovaciu schemu pre zarovnavanie dvoch DNA sekvencii (bez medzier). Mame dva modely, kazdy z nich vie vygenerovat 2 zarovnane sekvencie dlzky n.
Model R (random) reprezentuje nezavisle nahodne sekvencie
- Opat pouzijeme nase vrece s gulockami oznacenyni A,C,G,T, pricom gulocok oznacenych A je 30%, C 20%, G 20% a T 30%.
- Vytiahneme gulicku, zapiseme si pismeno, hodime ju naspat, zamiesame a opakujeme s dalsim pismenom atd az kym nevygenerujeme n pismen pre jednu sekvenciu a n pismen pre druhu
- Mame jednu sekvenciu ACT a druhu ACC. Aka je sanca, ze prave tieto sekvencie vygenerujeme v nasom modeli R?
- Nezavisle udalosti pre jednotlive pismena, t.j. Pr(X1=A)*Pr(X2=C)*Pr(X3=T)*Pr(Y1=A)*Pr(Y2=C)*Pr(Y3=C) = 0.3*0.2*0.3*0.3*0.2*0.2 = 0.000216
- Spolu mame v modeli
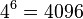 moznosti ako vygenerovat 2 DNA sekvencie dlzky 3
moznosti ako vygenerovat 2 DNA sekvencie dlzky 3
Model H (homolog) reprezentuje zarovnanie vzajomne suvisiacich sekvencii
- mame mech, v ktorom je napr.
- po 21% guliciek oznacenych AA, TT
- po 14% oznacenych CC, GG
- po 2.4% oznacenych AC, AG, CA, CT, GA, GT, TC, TG
- po 3.6% oznacenych AT, TA
- po 1.6% oznacenych CG, GC.
- Spolu mame 70% guliciek oznacenych rovnakymi pismenami, 30% roznymi
- n krat z mechu vytiahneme gulicku a pismena piseme ako stlpce zarovnania A1, A2,.., An.
- aka je pravdepodobnost, ze dostaneme ACT zarovnane s ACC?
- Pr(A1=AA)*Pr(A2=CC)*Pr(A3=TC) = 0.21*0.14*0.024 = 0.0007056
Skore zarovnania je log Pr(zarovnania v H)/Pr(zarovnania v R), t.j. log (0.0007056 / 0.000216) = 0.514105 (pre desiatkovy logaritmus)
- kladne skore znamena, ze model H lepsie zodpoveda datam (zarovnaniu) ako model R
- zaporne skore znamena, ze model R lepsie zodpoveda datam
Cvičenie pri počítači
- Stiahnite si súbor [7], uložte si ho a otvorte v Openoffice
- V záložke Matica vyplňte do žltej oblasti vzorce na výpočet pravdepodobnosti krátkeho zarovnania, logaritmus pomeru pravdepodobnosti a súčet skóre, pričom vo vzorcoch použijete odkazy na políčka v riadkoch 9-13, stĺpcoch B a E
- Súčet skóre by mal zhruba rovný desaťnásobku logaritmu pomeru - prečo vidíme rozdiely?
- Potom skúšajte meniť %GC a %identity v horných riadkoch tabuľky a pozrite sa, ako to ovplyvní skórovanie. Výsledné skóre zo stĺpca E ručne prepíšte (bez formúl) do tabuľky v záložke Výsledky. Prečo nastávaju také zmeny ako vidíte?
CI05
Jadrá s medzerami (spaced seeds)
- Prvá čast v prezentácii pdf
Vzorec na vypocet senzitivity jadra bez medzier
- Uvazujme jadro bez medzier dlzky w (ako v programe BLAST pre nukleotidy)
- Uvazujme pravdepodobnostny model zarovnania, v ktorom ma kazda pozicia pravdepodobnost p, ze bude zhoda a (1-p), ze bude nezhoda, medzery neuvazujeme.
- Nech f(L) je pravdepodobnost vyskytu jadra v zarovnani dlzky L, t.j. pravdepodobnost w za sebou iducich zhod.
-
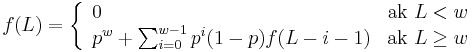
Vektorove jadra
- zarovnanie ako postupnost skore (napr 0/1 pre zhoda/nezhoda, male cele cisla pre BLOSUM63 maticu skorovania aminokyselin)
- Jadro je vektor vah v_1\dots v_k dlzky k a prahova hodnota T
- Okno zarovnania s_1\dots d_k je vyskyt jadra ak
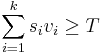
- Vyjadrite jadra s medzerami, BLAT-ove a BLASTp jadra ako vektorove jadra
Rychle hladanie jadier v sekvenciach
- Jadro bez medzier (BLAST)
- trivialna hash tabulka velkost 4^w
- Aho-Corasickovej algoritmus pre vsetky w-tice v prvom retazci
- sufixovy strom
- atd
- Jadro s medzerami
- Pouzijeme trivialnu hash tabulku, ale ukladame len znaky na neignorovanych poziciach
- Jadro BLAT-u
- Hash tabulka pre jednu sekvenciu, pre druhu hladame vsetky retazce v okoli danej k-tice ktore sa lisia najviac na jednom mieste (je ich 3k+1)
- Ak by sme dovolili viac rozdielov, ich pocet by rychlo narastal
Senzitivita jadra s medzerami
- Da sa rekurentny vzorec vyssie rozsirit na jadro s medzerami?
-
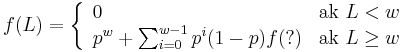
- Co by sme pouzili namiesto otaznika?
- Napr pre jadro 1011 a zaciatok zarovnania 1?10 moze byt vyskyt na pozicii 3, ale jeho pravdepodobnost nie je f(L-2), lebo mame dane prve dva znaky
Vseobecny algoritmus pre jadra s medzerami/vektorove jadra
- Zial vo vseobecnosti exponencialny od dlzky jadra, ale pre male jadra sa da zbehnut v rozumnom case
- Zarovnanie si predstavme ako nejaku postupnost nad abecedou D (napr D={z,h} kde z je zhoda, h nezhoda). Nech q_d je pravdepodobnost znaku d z D v zarovnani
- Uvazujme jadro dlzky k, co si vieme predstavit ako nejake pravidlo, ktore urci, ci dany retazec dlzky k nad D je alebo nie je vyskytom jadra
- H nech je mnozina vsetkych vyskytov jadra (t.j. mnozina retazcov x dlzky k nad mnozinou D, ktore splnaju podmienku jadra)
- Nech f(L,x) je pravdepodobnost vyskytu jadra v zarovnani dlzky L, ktore zacina na retazec x, kde x je hocijaky retazec nad D dlzky najviac k
-
![f(L,x)=\left\{{\begin{array}{ll}0&{\mbox{ak }}L<k\\1&{\mbox{ak }}L\geq k\wedge x\in H\\f(L-1,x[2\dots k])&{\mbox{ak }}L\geq k\wedge x\notin H\wedge |x|=k\\\sum _{{d\in D}}q_{d}f(L,xd)&{\mbox{ak }}L\geq k\wedge |x|<k\\\end{array}}\right.](/vyuka/mbi/images/math/9/e/1/9e1d669be9402a15f0f9b57c8165defa.png)
- f(L,x) budeme pocitat pre rastuce L a pre kazde L od najdlhsich x.
- vysledok je
Rychlejsi algoritmus
- Nech H_p je mnozina prefixov retazcov z H (mozne vyskyty) a H_g je mnozina prefixov retazcov z H, ktorych kazde rozsirenie na dlzku k je vyskyt (garantovane vyskyty)
-
- Nech f(L,x) je pravdepodobnost vyskytu jadra v zarovnani dlzky L, ktore zacina na retazec x.
-
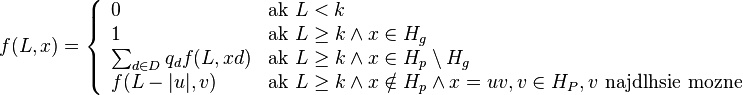
- f(L,x) budeme pocitat pre rastuce L a pre kazde L od najdlhsich x. Staci uvazovat x z H_p alebo x, ktore dostaneme pridanim jedneho znaku k slovu z H_p.
- vysledok je
- Pre jadro s g medzerami
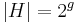 a
a 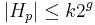 .
.
Počítanie fylogenetických stromov
- Ako definujeme strom v teorii grafov? suvisly acyklicky neorientovany graf
- Strom s n vrcholmi ma n-1 hran
- Nezakoreneny binarny fylogeneticky strom: neorientovany suvisly acyklicky graf, v listoch sucasne druhy, vsetky vnutorne vrcholy stupna 3
- Zakoreneny binarny fylogeneticky strom: vsetky hrany orientujeme od korena smerom k listom, kazdy vnutorny vrchol ma dve deti
- Niekedy uvazujeme aj nebinarne stromy, v ktorych mame vnutorne vrcholy vyssieho stupna
- Zakoreneny binarny strom s n listami ma n-1 vnutornych vrcholov, teda 2n-2 hran
- Nezakoreneny binarny strom s n listami ma n-2 vnutornych vrcholov, teda 2n-3 hran
- Pocet nezakorenenych fylogenetickych stromov s n listami:
- a(3) = 1, a(4) = 3, a(n+1) = a(n) * (2n-3) a teda a(n) = 1 * 3 * 5 * ... * (2n-5) = (2n-5)!!
- Pocet zakorenenych fylogenetickych stromov s n listami:
- zakoren strom s n listami kazdy 2n-3 sposobmi, teda (2n-3)!!
CB05
Príklady stavových automatov pre HMM
Uvazujme HMM so specialnym zaciatocnym stavom b a koncovym stavom e, ktore nic negeneruju.
- Nakreslite HMM (stavovy diagram), ktory generuje sekvencie, ktore zacinaju niekolkymi cervenymi pismenami a potom obsahuju niekolko modrych
- Ako treba zmenit HMM, aby dovoloval ako "niekolko" aj nula?
- Ako treba zmenit HMM, aby pocet cervenych aj modrych bol vzdy parne cislo?
- Ako zmenit HMM, aby sa striedali cervene a modre kusy parnej dlzky?
V dalsich prikladoch uvazujeme aj to, ktore pismena su v ktorom stave povolene (pravdepodobnost emisie > 0) a ktore su zakazane
- cervena sekvencia dlzky dva, ktora zacina na A
- cervena sekvencia dlzky dva, ktora je hocico okrem AA
- toto sa da rozsirit na HMM, ktory reprezentuje ORF, teda nieco, co zacina start kodonom, potom niekolko beznych kodonov, ktore nie su stop kodonom a na koniec stop kodon
Dalsi biologicky priklad HMM: topologia transmembranovych proteinov.
E-hodnota (E-value) zarovnania
- Priklady k tejto casti v prezentacii pdf
- Mame dotaz dlzky m, databazu dlzky n, skore najlepsieho lokálneho zarovnania S
- E-value je ocakavany pocet zarovnani so skore aspon S ak dotaz aj databaza su nahodne
- Hrackarsky priklad: dotaz dlzky m=4, databaza dlzky n=200, S=4, t.j. presny vyskyt dotazu v databaze (pre presne vyskyty sa pravdepodobnosti pocitaju ovela lahsie ako ked dovolime nezhody a medzery)
- Zoberme nas nahodny model s obsahom GC 40%
- Mame vrece s gulockami oznacenymi A,C,G,T, pricom gulocok oznacenych A je 30%, C 20%, G 20% a T 30%.
- Vytiahneme gulicku, zapiseme si pismeno, hodime ju naspat, zamiesame a opakujeme s dalsim pismenom atd az kym nevygenerujeme m pismen pre dotaz a n pismen pre databazu
- Pre nase vygenerovane sekvencie spocitame, kolkokrat sa dotaz vyskytuje v databaze
- Cely experiment opakujeme vela krat a spocitame priemerny pocet vyskytov, co bude odhad E-value
Presny vypocet E-value pre jednoduchy pripad, ked sa dotaz vyskytuje v databaze bez jedinej zmeny (uvedene pre zaujimavost, nerobili sme):
- Podobny vypocet sme videli na cviceniach 2
- Mame dotaz X1X2...Xm a databazu Y1Y2...Yn. Aka je sanca, ze X sa nachadza na zaciatku Y, t.j. ze X1=Y1, X2=Y2... Xm=Ym?
- Pripominame, ze pravdepodobnost zhody dvoch nukleotidov napr X1 a Y1 je Pr(X1=Y1) = Pr(X1=Y1=A) + Pr(X1=Y1=C) + Pr(X1=Y1=G) + Pr(X1=Y1=T) = 0.3*0.3+0.2*0.2+0.2*0.2+0.3*0.3 = 0.26.
- Pre rozne pozicie v X mame nezavisle udalosti, t.j. pravdepodobnost vyskytu X na prvej pozicii je Pr(X1=Y1)*Pr(X2=Y2)*...*Pr(Xm=Ym) = 0.26 * 0.26 * ... * 0.26 = 0.26^m (0.26 na m-tu)
- Tak isto nam vyjde aj pravdepodobnost vyskytu X na hociktorej inej pevnej pozicii v Y
- Pravdepodobnost, ze sa X nachadza na aspon jednej pozicii v Y (t.j. P-value zarovnania) je tazsie zratat - moze sa vyskytovat aj viackrat, udalosti ze sa nachadza na pozicii i sa navzajom nevylucuju
- Ale priemerny pocet vyskytov na pozicii i je 0.26^m, mame n-m+1 pozicii zaciatku, takze celkovy ocakavany pocet vyskytov je (n-m+1)0.26^m
- Toto je stredna hodnota nahodnej premennej C, ktora oznacuje pocet presnych vyskytov
- E(C) = E(C_1)+E(C_2)+...E(C_(n-m+1)), kde C_i je nahodna premenna, ktora je 1 ak sa X nachadza na pozicii i v Y
- E(C_i) = 0*Pr(C_i=0)+1*Pr(C_i=1) = Pr(C_i=1) = 0.26^m
- Ak je n velke v porovnani s m, clen -m+1 mozeme zanedbat, t.j. mame zhruba
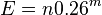
- Ako sa meni toto cislo s velkostou databazy n? linearne rastie (zdvojnasobime databazu, zdvojnasobi sa E-value)
- Ako sa meni s S=m? Exponencialne klesa, t.j. ak predlzime dotaz o 1 (a stale dostaneme skore m), E-value klesne zhruba na stvrtinu
- Ako závisí od GC obsahu? - to je zakódované v čísle 0.26 (pravdepodbnosť zhody dvoch nezávislých báz). Pre GC 50% je táto pravdepodobnosť 0.25 a pre GC 100% je 0.5.
Stredna hodnota pre vseobecny pripad lokalneho zarovnania so skore S (nemusi sa cely dotaz nachadzat bez zmeny v databaze)
- zlozita matematicka teoria [8]
- E-value sa priblizne da odhadnut vzorcom:
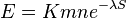
- n a m su dlzky porovnavanych sekvencii, S je skore, K a lambda su parametre, ktore zavisia od skorovacej schemy a od frekvencii vyskytu jednotlivych baz v nasom modeli nahodnej sekvencie.
- Napr blastn pre skorovaci system zhoda 1, nezhoda -1, medzera -2 používa lambda=0.800, K=0.0640
-
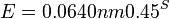 .
.
- Opäť zdvojnásobenie dĺžky databázy alebo dĺžky dotazu zdvojnásobí E-value
- Zníženie skóre o 1 tiež zhruba zdvojnásobí E-value (nasobenie 2.2)
- Cislo, ktorym nasobime, zalezi od lambda a teda od skorovacej schemy a frekvencii vyskytu baz
Hľadanie génov v prokaryotických genómoch
K hladaniu genov a komparativnej genomike pozri aj prezentaciu pdf
- ORF: open reading frame, jednoduche hladanie
- ako najst zaciatok, ako rozlisit psedogeny a nahodne ORF-y
- samotrenujuce sa HMM, codon bias, GC%
E. coli http://nar.oxfordjournals.org/content/34/1/1.full
- Prvykrat sekvenovana a anotovana 1997
- Porovnanie s verziou 2005 (oprava sekvenovacích chýb aj chýb v anotácii)
- 682 zmien v start kodone
- 31 génov zrušených
- 48 nových génov
- Celkovo asi 4464 génov
Programy na anotovanie prokaryotických genómov
Histónové modifikácie
- A. Barski, S. Cuddapah, K. Cui, T. Roh, D. Schones, Z. Wang, G. Wei, I. Chepelev, K. Zhao (2007) High-Resolution Profiling of Histone Methylations in the Human Genome Cell, Volume 129, Issue 4, Pages 823-837 pdf
Gény v ľudskom genóme
- What is a gene, post-ENCODE? History and updated definition. Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, Emanuelsson O, Zhang ZD, Weissman S, Snyder M.
- Most "dark matter" transcripts are associated with known genes. H Van Bakel, C Nislow, BJ Blencowe, TR Hughes - PLoS Biol, 2010
- Transcribed dark matter: meaning or myth? CP Ponting, TG Belgard - Human molecular genetics, 2010
- Landscape of transcription in human cells. Djebali et al (ENCODE), Nature 2012
CI06
HMM opakovanie
Parametre HMM:
-
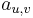 : prechodová pravdepodobnosť zo stavu
: prechodová pravdepodobnosť zo stavu 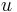 do stavu
do stavu 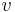
-
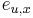 : pravdepodobnosť emisie
: pravdepodobnosť emisie 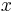 v stave
v stave
-
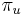 : pravdepodobnosť, že začneme v stave
: pravdepodobnosť, že začneme v stave
- Sekvencia
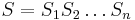
- Anotácia
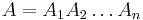
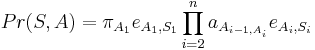
Trénovanie
- Proces, pri ktorom sa snažíme čo najlepšie odhadnúť pravdepodobnosti a v modeli podľa trénovacích dát
Usudzovanie (inferencia)
- Proces, pri ktorom sa snažíme pre sekvenciu
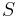 nájsť anotáciu
nájsť anotáciu 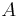 , ktorá sekvenciu emituje s veľkou pravdepodobnosťou.
, ktorá sekvenciu emituje s veľkou pravdepodobnosťou.
Inferencia pomocou najpravdepodobnejšej cesty, Viterbiho algoritmus
Hľadáme najpravdepodobnejšiu postupnosť stavov , teda 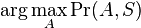 . Úlohu budeme riešiť dynamickým programovaním.
. Úlohu budeme riešiť dynamickým programovaním.
- Podproblém
![V[i,u]](/vyuka/mbi/images/math/8/2/b/82b63b00ee9d341b172ed7a0c7c44f7b.png) : Pravdepodobnosť najpravdepodobnejšej cesty končiacej po
: Pravdepodobnosť najpravdepodobnejšej cesty končiacej po 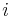 krokoch v stave , pričom vygeneruje
krokoch v stave , pričom vygeneruje 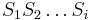 .
.
- Rekurencia:
-
![V[1,u]=\pi _{u}e_{{u,S_{1}}}](/vyuka/mbi/images/math/f/1/3/f13d26742a6a93123093e174c3fd90aa.png) (*)
(*)
-
![V[i,u]=\max _{w}V[i-1,w]a_{{w,u}}e_{{u,S_{i}}}](/vyuka/mbi/images/math/e/3/b/e3bf534bb50c39843ed166123ecc374b.png) (**)
(**)
-
Algoritmus:
- Nainicializuj
![V[1,*]](/vyuka/mbi/images/math/5/d/9/5d9f1d4ebd3080a0804a6501977b5dae.png) podľa (*)
podľa (*)
- for i=2 to n=dĺžka reťazca
- for u=1 to m=počet stavov
- vypočítaj pomocou (**)
- vypočítaj
- for u=1 to m=počet stavov
- Maximálne
![V[n,j]](/vyuka/mbi/images/math/1/2/9/12924883f1d662364326a1d3f0fed497.png) je pravdepodobnosť najpravdepodobnejšej cesty
je pravdepodobnosť najpravdepodobnejšej cesty
Aby sme vypísali anotáciu, pamätáme si pre každé stav 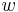 , ktorý viedol k maximálnej hodnote vo vzorci (**).
, ktorý viedol k maximálnej hodnote vo vzorci (**).
Zložitosť: 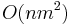
Poznámka: pre dlhé sekvencie budú čísla veľmi malé a môže dôjsť k podtečeniu. V praxi teda používame zlogaritmované hodnoty, namiesto násobenia súčet.
Inferencia - dopredný algoritmus
Aká je celková pravdepodobnosť, že vygenerujeme sekvenciu , t.j. 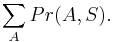 Podobný algoritmus ako Viterbiho.
Podobný algoritmus ako Viterbiho.
Podproblém ![F[i,u]](/vyuka/mbi/images/math/4/e/6/4e6962ec814d01e4ca0d1f2aa085a831.png) : pravdepodobnosť, že po krokoch vygenerujeme
: pravdepodobnosť, že po krokoch vygenerujeme 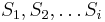 a dostaneme sa do stavu .
a dostaneme sa do stavu .
![F[i,u]=\Pr(A_{i}=u\wedge S_{1},S_{2},\dots ,S_{i})=\sum _{{A_{1},A_{2},\dots ,A_{i}=u}}\Pr(A_{1},A_{2},...,A_{i}\wedge S_{1},S_{2},...,S_{i})](/vyuka/mbi/images/math/9/c/5/9c51082607764a6c99095ebaa1ba26f4.png)
![F[1,u]=\pi _{u}e_{{u,S_{1}}}](/vyuka/mbi/images/math/f/e/a/fead9f93f8bf3927b34f49597d884996.png)
![F[i,u]=\sum _{v}F[i-1,v]a_{{v,u}}e_{{u,S_{i}}}](/vyuka/mbi/images/math/d/1/c/d1c9473aeb8cc364771bc9c575c7a73f.png)
Celková pravdepodobnosť ![\sum _{u}F[n,u]](/vyuka/mbi/images/math/1/b/2/1b2108be9d84620e173cb861e49c6b35.png)
Inferencia - posterior decoding
Aposteriórna pravdepodobnosť stavu u na pozícii i: 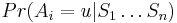
Pre každý index i chceme nájsť stav u s najväčšiou aposteriórnou pravdepodobnosťou, dostaneme tak inú možnú anotáciu.
Spustíme dopredný algoritmus a jeho symetrickú verziu, spätný algoritmus, ktorý počíta hodnoty
![B[i,u]=\Pr(A_{i}=u\wedge S_{{i+1}}\dots S_{n})](/vyuka/mbi/images/math/6/e/8/6e82648bde89df23b004629c06c05c23.png)
Aposteriórna pravdepodobnosť stavu u na pozícii i: ![Pr(A_{i}=u|S_{1}\dots S_{n})=F[i,u]B[i,u]/\sum _{u}F[n,u].](/vyuka/mbi/images/math/2/7/c/27cea5da3d3a25dfb8a677a41dc9e9b1.png)
Posterior decoding uvažuje všetky anotácie, nielen jednu s najvyššou pravdepodobnosťou. Môže však vypísať anotáciu, ktorá má sama o sebe nulovú pravdepodobnosť (napr. počet kódujúcich báz v géne nie je deliteľný 3).
Trénovanie HMM
- Stavový priestor + povolené prechody väčšinou ručne
- Parametre (pravdepodobnosti prechodu, emisie a počiatočné) automaticky z trénovacích sekvencií
- Ak máme anotované trénovacie sekvencie, jednoducho počítame frekvencie
- Ak máme iba neanotované sekvencie, snažíme sa maximalizovať vierohodnosť trénovacích dát v modeli. Používajú sa heuristické iteratívne algoritmy, napr. Baum-Welchov, ktorý je verziou všeobecnejšieho algoritmu EM (expectation maximization).
- Čím zložitejší model a viac parametrov máme, tým potrebujeme viac trénovacích dát, aby nedošlo k preučeniu, t.j. k situácii, keď model dobre zodpovedá nejakým zvláštnostiam trénovacích dát, nie však ďalším dátam.
- Presnosť modelu testujeme na zvláštnych testovacích dátach, ktoré sme nepoužili na trénovanie.
Tvorba stavového priestoru modelu
- Promótor + niekoľko prokaryotických génov
- Repeaty v intrónoch: multiple path problem
- Intrón má dĺžku aspoň 10
Zovšeobecnené HMM
- Problém s modelovaním rozdelenia dĺžok - v základnom modeli je geometrické
- Zovseobecnene HMM v jednom stave vygenerujú viac znakov
- Viterbi alg. a spol budú pomalšie (kvadratické od dĺžky sekvencie)
Párové HMM (pair HMM)
Nebrali sme, uvedene pre zaujimavost
- Emituje dve sekvencie
- V jednom kroku moze emitovat:
- pismenka v oboch sekvenciach naraz
- pismenko v jednej skevencii
- pismenko v druhej sekvencii
Priklad: HMM s jednym stavom v, takym, ze
-
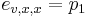
-
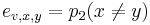 ,
,
-
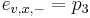 ,
,
-
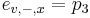
- tak, aby sucet emisnych pravdepodobnosti bol 1
- Co reprezentuje najpravdepodobnejsia cesta v tomto HMM?
Zlozitejsi HMM: tri stavy M, X, Y, uplne navzajom poprepajane
-
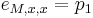
-
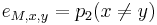 ,
,
-
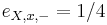 ,
,
-
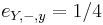 ,
,
- Co reprezentuje najpravdepodobnejsia cesta v tomto HMM?
Viterbiho algoritmus pre parove HMM
- V[i,j,u] = pravdepodobnost najpravdepodobnejsej postupnosti stavov, ktora vygeneruje x1..xi a y1..yj a skonci v stave u
-
![V[i,j,u]=\max _{w}\left\{{\begin{array}{l}V[i-1,j-1,w]\cdot a_{{w,u}}\cdot e_{{u,x_{i},y_{j}}}\\V[i-1,j,w]\cdot a_{{w,u}}\cdot e_{{u,x_{i},-}}\\V[i,j-1,w]\cdot a_{{w,u}}\cdot e_{{u,-,y_{j}}}\\\end{array}}\right.](/vyuka/mbi/images/math/d/5/0/d5064f1dc674bec18ec3404260675e81.png)
- Casova zlozitost O(mnk^2) kde m a n su dlzky vstupnych sekvencii, k je pocet stavov
Ako by sme spravili parove HMM na hladanie genov v dvoch sekvenciach naraz?
- Predpokladajme rovnaky pocet exonov
- V exonoch medzery len cele kodony (oboje zjednodusuje)
- Inde hocijake medzery
CB06
Fylogenetické stromy
Terminológia:
- zakorenený strom, rooted tree
- nezakorenený strom, unrooted tree
- hrana, vetva, edge, branch
- vrchol, uzol, vertex, node
- list, leaf, leaf node, tip, terminal node
- vnútorný vrchol, internal node
- koreň, root
- podstrom, subtree, clade
Zopár faktov o stromoch
- Majme zakorenený strom s n listami, v ktorom má každý vnútorný vrchol 2 deti. Takýto strom vždy má n-1 vnútorných vrcholov a 2n-2 vetiev (prečo?)
- Majme nezakorenený strom s n listami, v ktorom má každý vnútorný vrchol 3 susedov. Takýto strom vždy má n-2 vnútorných vrcholov a 2n-3 vetiev
- Koľkými spôsobmi môžeme zakoreniť nezakorenený strom s n listami?
- koreň môže byť na hociktorej vetve stromu, teda je 2n-3 možností zakorenenia
- Ak nakreslíme zakorenený strom obvyklým spôsobom, listy sú usporiadané zhora nadol (alebo zľava doprava). Koľko rôznych poradí listov vieme dostať rôznym zakresľovaním toho istého stromu s n listami?
- máme n-1 vnutornych vrcholov, v kazdom mozeme vymenit lave a prave dieta. Pre kazdu konfiguraciu takychto vymen dostavame ine poradie, celkovy pocet poradi je 2n-1
- Koľko je vôbec roznych poradí listov, ak neberieme do úvahy strom?
- je to n!, t.j. sucin cisel od 1 po n. Napr. pre n=4 mame 4! = 1*2*3*4 = 24 roznych poradi n listov, ale len 23=8 z nich zodpoveda danemu stromu
- Čo vieme zistit o pribuznosti organizmov z nezakoreneneho stromu (napr. kvartet 4 organizmov)?
- skusime zakorenit vsetkymi sposobbmi a vidime, ze o ziadnych dvoch listoch nevieme povedat, ze by boli sesterske (evolucne blizsie nez ostatne), lebo koren stromu moze byt zrovna na niektorej hrane veducej ku nim
- vieme vsak zistit, ze niektore dvojice sesterske nebudu
Ine pouzitie stromov v informatike:
- uvidime hierarchicke zhlukovanie, bayesovske siete, ale tiez efektivne datove struktury
Bootstrap
- Náhodne vyberieme niektoré stĺpce zarovnania, zostrojíme strom vybranou metódou
- Celé to opakujeme veľa krát
- Značíme si, koľkokrát sa ktorá vetva opakuje v stromoch, ktoré dostávame
- Pri nezakorenených stromoch je vetva rozdelenie listov na dve skupiny (bipartícia)
- Nakoniec zostavíme strom z celých dát a pozrieme sa ako často sa ktorá jeho vetva vyskytovala
- Môžeme zostavit aj strom z často sa vyskytujúcich hrán (napr. tých, ktoré sú vo viac ako 50% stromov)
- Bootstrap hodnoty nám dajú určitý odhad spoľahlivosti, hlavne ak máme celkovo málo dát (krátke zarovnanie)
- Ak však dáta nezodpovedajú vybranej metóde/modelu, tak aj pre zlý strom môžeme dostať vysoký bootstrap
Príklad

- Robili sme 100x bootstrap, 40x sme dostali strom (i) na obrázku, 40x sme dostali strom (ii) a 20x sme dostali strom (iii)
- Strom (iii) sme dostali aj spustením metody na celých dátach
- Zistite úroveň bootstrap podpory pre jednotlivé vetvy stromu (iii)
- Ktoré ďalšie vetvy majú podporu aspoň 20%?
- Aký strom by sme dostali, ak by sme chceli nechať iba vetvy s podporou aspoň 80%?
Opakovanie pravdepodobnostných modelov
Keď počítame pravdepodobnosť, rozmýšľame o myšlienkovom experimente, v ktorom hádžeme kockou, ťaháme gulôčky z vreca a pod.
- Dôležité je vždy si poriadne uvedomiť, ako tento experiment prebieha
- Tieto myšlienkové experimenty však nastavujeme tak, aby odzrkadľovali nejaké aspekty reality, napr. skutočných DNA sekvencií, ich evolúcie a pod.
- Takže pravdepodobnosti, ktoré spočítame v idealizovanom svete nám možno niečo povedia o reálnom svete
- Slávny citát štatistika Georga Boxa "All models are wrong, but some are useful."
Aké sme doteraz videli modely
- Skórovacie matice: porovnavame model nahodnych sekvencii a model nahodnych zarovnani
- E-value v BLASTe: nahodne vygenerujeme databazu a dotaz (query), kolko bude v priemere medzi nimi lokalnych zarovnani so skore aspon T?
- Hladanie genov: model generujuci sekvenciu+anotaciu naraz (parametre nastavene na znamych genoch). Pre danu sekvenciu, ktora anotacia je najpravdepodobnejsia?
- Evolucia, Jukes-Cantorov model: model generujuci stlpec zarovnania. Nezname parametre: strom, dlzky hran. Pre danu sadu stlpcov zarovnania, ktore parametre povedu k najvacsej pravdepodobnosti?
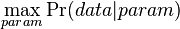
- Trochu detailov: pravdepodobnost zmeny/nezmeny na hrane dlzky t:
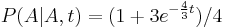 ,
, 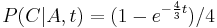
- Ak pozname ancestralne sekvencie, vieme spocitat pravdepodobnost dat
- Ancestralne sekvencie su nahodne premenne, ktore nas nezaujimaju: marginalizujeme ich (uvazujeme vsetky ich mozne hodnoty)
- Trochu detailov: pravdepodobnost zmeny/nezmeny na hrane dlzky t:
Zložitejšie evolučné modely
- Jukes-Cantorov model uvažuje len dĺžku hrany udanú ako priemerný počet substitúcií (vrátane tých, ktore nevidíme, kvôli tomu, že boli dve na tom istom mieste)
- Nie všetky substitúcie sa dejú rovnako často: tranzície (v rámci pyrimidínov T<->C, v rámci purínov A<->G) sú pravdepodobnejšie ako tranzverzie (A,G)<->(C,T)
- Nie všetky nukleotidy sa v danom genóme vyskytujú rovnako často (napr. mitochondriálne genómy majú nízky obsah GC)
- Tieto javy zachytava HKY model (Hasegawa, Kishino & Yano)
- Matica rychlosti (substitution rate matrix)
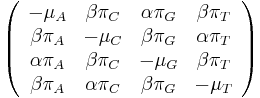
-
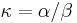 je pomer rychlosti, ktorymi sa deju tranzicie vs. transverzie
je pomer rychlosti, ktorymi sa deju tranzicie vs. transverzie
-
 je frekvencia bazy
je frekvencia bazy  v sekvencii
v sekvencii
- Rychlost, ako sa deje substitucia z X do Y je sucin pravdepodobnosti Y a faktoru, ktory zavisi od toho, ci ide o tranziciu alebo transverziu
- Sucet kazdeho riadku matice ma byt 0, t.j.
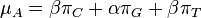
- Matica sa znormalizuje tak, aby priemerny pocet substitucii za jednotku casu bol 1
- Matica ma styri parametre:
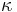 a tri frekvencie (stvrta musi doplnit do 1) plus dlzka hrany
a tri frekvencie (stvrta musi doplnit do 1) plus dlzka hrany
- Zlozitejsi model lepsie zodpoveda skutocnym procesom, ale na odhad viac parametrov potrebujeme viacej dat.
- Existuju metody, ktore pre dany cas t z matice rychlosti spocitaju pravdepodobnost, ze baza X zmutuje na bazu Y Pr(Y|X,t)
- Napr. pre velmi maly cas
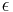 mame
mame  je zhruba
je zhruba 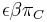
- Pre rozumne dlhe casy toto neplati, preto sa pouzivaju algebraicke metody, ktore beru do uvahy moznost viacerych substitucii na tom istom mieste
- Je aj vela inych modelov s mensim alebo vacsim poctom parametrov
Praktická ukážka tvorby stromov
- V UCSC browseri mozeme ziskavat viacnasobne zarovnania jednotlivych genov (nukleotidy alebo proteiny). Nasledujuci postup nemusite robit, subor si stiahnite tu: http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb06/cb06-aln.fa
- UCSC browseri si pozrieme usek ludskeho genomu (verzia hg38) chr6:135,851,998-136,191,840 s genom PDE7B (phosphodiesterase 7B) [13]
- Na modrej liste zvolime Tools, Table browser. V nastaveniach tabuliek Group: Genes and Gene Predictions, Track: GENCODE v 24., zaklikneme Region: position, a Output fomat: CDS FASTA alignment a stlacime Get output
- Na dalsej obrazovke zaklikneme show nucleotides. zvolime MAF table multiz100way a vyberime si, ktore organizmy chceme. V nasom pripade z primatov zvolime chimp, rhesus, bushbaby, z inych cicavcov mouse, rat, rabbit, pig, cow, dog, elephant a z dalsich organizmov opposum, platypus, chicken, stlacime Get output.
- Vystup ulozime do suboru, nechame si iba prvu formu genu (uc003qgp.4), z mien sekvencii zmazeme spolocny zaciatok (uc003qgp.4), pripadne celkovo prepiseme mena na anglicke nazvy
- Skusme zostavit strom na stranke http://www.phylogeny.fr/alacarte.cgi
- Z krokov spracovania zrusime Multiple alignment (sekvencie mame zarovnane) a Alignment curation
- Z algoritmov na tvorbu stromu zvolime ProtDist/FastDist + BioNJ, t.j. metodu spajania susedov (mozete si skusit aj ine)
- Vizualizaciu nechajte TreeDyn
- Na dalsej obrazovke
- zadame do okienka zarovnanie uvedene vyssie
- v nastaveniach BioNJ zvolime 100 bootstrapov, Jukes-Cantor model (mozet skusit aj Kimura 2-parameter)
- Ak by sme pouzili namiesto BioNJ program PhyML (metoda maximalnej vierohodnosti), mozeme nastavovat viacero parametrov modelu
- Vo vyslednom strome mozeme zmenit zakorenenie, aby sme mali sliepku (chicken) ako outgroup
- Vysledok s povodnym zakorenenim
- Vysledok so spravnym zakorenenim (chicken = outgroup)
- "Spravny strom" [14] v nastaveniach Conservation track-u v UCSC browseri (podla clanku Murphy WJ et al Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001 [15])
- Nas strom ma dost zlych hran: zle postavenei hlodavcov, ale aj slona a psa. Zle postavenie hlodavcov moze byt sposobene long branch attraction.
- Na serveri sa daju skusit spravit aj zarovnania, vtedy treba zacat z nezarovnanych sekvencii: [16]
CI07
Substitucne modely - odvodenie
- Nech
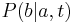 je pravdepodobnosť, že ak začneme s bázou a, tak po čase t budeme mať bázu b.
je pravdepodobnosť, že ak začneme s bázou a, tak po čase t budeme mať bázu b.
- Pre dane t mozeme take pravdepodobnosti usporiadat do matice 4x4 (ak studujeme DNA):
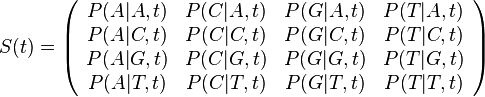
- Riadky zodpovedaju povodnej baze a, stlpce novej baze b
Poziadavky na S(t)
- Intuitivne cim vacsie t, tym vacsia pravdepodobnost zmeny, pre nulovy cas este ziadna zmena nemala kedy nastat, mame teda
 (jednotkova matica)
(jednotkova matica)
- Naopak ked t ide do nekonecna, kazda baza velakrat zmutovala a teda uz prilis nezalezi, co to bolo na zaciatku. S(t) ma teda v limite pre velke t vsetky riadky rovnake.
-
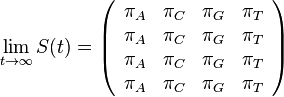
- Rozdelenie pravdepodobnosti
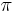 nazyvame limitne (equilibrium)
nazyvame limitne (equilibrium)
- Predpokladame tiez, ze pravdepodobnost mutacie zavisi len od aktualnej bazy, nie od minulych stavov a ze charakter procesu mutacii sa v case nemeni. Teda ak mame matice pre casy
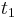 a
a 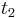 , vieme spocitat maticu pre cas
, vieme spocitat maticu pre cas 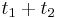 :
: 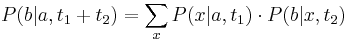 a teda v maticovej notacii
a teda v maticovej notacii  . Preto takyto model nazyvame multiplikativny.
. Preto takyto model nazyvame multiplikativny.
- Ak by sme uvazovali iba diskretne (celocislene) casy, stacilo by nam urcit iba
 a vsetky ostatne casy dostaneme umocnenim tejto matice. Je vsak elegantnejsie mat
a vsetky ostatne casy dostaneme umocnenim tejto matice. Je vsak elegantnejsie mat 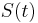 definovane aj pre realne t.
definovane aj pre realne t.
- Tento typ modelu sa nazyva Markovov retazec so spojitym casom (continuous-time Markov chain)
Jukes-Cantorov substitucny model
- Tento model predpoklada, ze vsetky substitucie su rovnako pravdepodobne, matica teda musí vyzerať nejako takto:
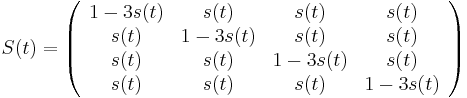
Matica rychlosti pre J-C model
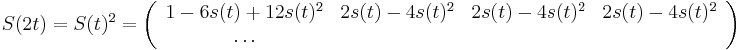
- Pre velmi maly cas t je s(t) velmi male cislo (blizke 0) a pre velmi male s(t) su kvadraticke cleny
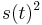 ovela mensie ako linearne cleny s(t) a teda
ovela mensie ako linearne cleny s(t) a teda
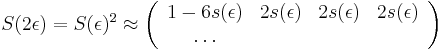
- Aj pre ine rozumne male nasobky
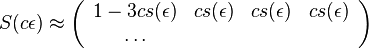
- Tento vztah dava zmysel: pre velmi male t mozeme zanedbat moznost, ze baza zmutovala viackrat a pravdepodobnost jednej mutacie linearne rastie s dlzkou casu.
- Ak oznacime
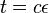 a
a 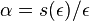 dostaneme pre velmi male t
dostaneme pre velmi male t
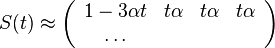
- Vytvorme si teraz maticu rychlosti (intenzit) (transition rate matrix, substitution rate matrix)
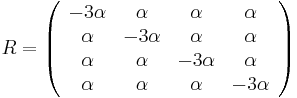
- Dostavame, ze pre velmi male casy plati
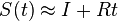
-
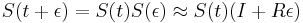 a teda
a teda 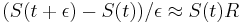
- V limite dostaneme
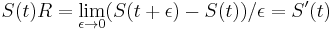 (diferencialne rovnice, pociatocny stav ).
(diferencialne rovnice, pociatocny stav ).
- Nasobenim matic S(t) a R dostavame, ze diagonalny prvok
 je
je  a nediagonalny
a nediagonalny 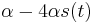 . Takze dostavame diferencialnu rovnicu
. Takze dostavame diferencialnu rovnicu 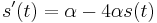 z rovnosti nediagonalnych prvkov (z rovnosti diagonalnych prvkov dostavame tu istu rovnicu len prenasobenu konstantou -3).
z rovnosti nediagonalnych prvkov (z rovnosti diagonalnych prvkov dostavame tu istu rovnicu len prenasobenu konstantou -3).
Poriadnejsie odvodenie diferencialnej rovnice
-
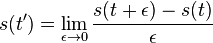 z definicie limity
z definicie limity
-
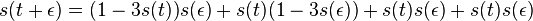 podla multiplikativnosti S(t)
podla multiplikativnosti S(t)
-
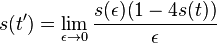 po uprave
po uprave
-
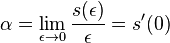 (oznacenie)
(oznacenie)
-
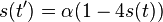 (finalna diferencialna rovnica, rovnaka ako predtym pre matice)
(finalna diferencialna rovnica, rovnaka ako predtym pre matice)
- Riesenie diferencialnej rovnice
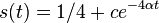 pre kazdu konstantu c
pre kazdu konstantu c
- Mozeme overit dosadenim do rovnice, pricom
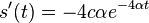
- c=-1/4 dopocitame z pociatocnej podmienky s(0)=0
- overime tiez, ze
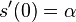
Vlastnosti riesenia
- Takze mame maticu:
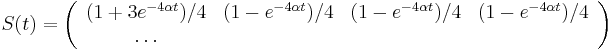
- Ked
 , dostavame r(t)=s(t)=1/4.
, dostavame r(t)=s(t)=1/4.
-
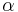 je teda pravdepodobnost konkretnej zmeny za jednotku casu, ak uvazujeme velmi kratke casy alebo presnejsie derivacia prvku s(t) vzhladom na t v bode 0
je teda pravdepodobnost konkretnej zmeny za jednotku casu, ak uvazujeme velmi kratke casy alebo presnejsie derivacia prvku s(t) vzhladom na t v bode 0
- Aby sme nemali naraz aj aj
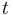 , zvykneme maticu R normalizovat tak, aby priemerny pocet substitucii za jednotku casu bol 1. V pripade Jukes-Cantorovho modelu je to ked
, zvykneme maticu R normalizovat tak, aby priemerny pocet substitucii za jednotku casu bol 1. V pripade Jukes-Cantorovho modelu je to ked 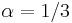 .
.
Pouzitie na odhad evolucnej vzdialenosti
- V case je pravdepodobnost, ze uvidime zmenenu bazu
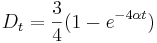
- V realnom pouziti (vypocet matice vzdialenosti pre metodu spajania susedov) mame dve zarovnane sekvencie, medzi ktorymi vidime
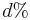 zmenenych baz, chceme odhadnut t
zmenenych baz, chceme odhadnut t
- Spatne teda zratame t, ktore by hodnote
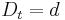 prinalezalo.
prinalezalo.
- Spatne teda zratame t, ktore by hodnote
- Dostavame teda vzorec pre vzdialenost, ktory sme videli na prednaske
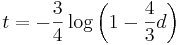
- Ak
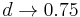 , dostavame
, dostavame
- Preco sme ten vzorec odvodili takto? V skutocnosti chceme najst najvierohodnejsiu hodnotu t, t.j. taku, pre ktore hodnota P(data|t) bude najvacsia. Zhodou okolnosti vyjde takto.
Substitučné matice zhrnutie
- S(t): matica 4x4, kde políčko
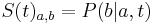 je pravdepodobnosť, že ak začneme s bázou a, tak po case t budeme mať bázu b.
je pravdepodobnosť, že ak začneme s bázou a, tak po case t budeme mať bázu b.
- Jukes-Cantorov model predpokladá, že táto pravdepodobnosť je rovnaká pre každé dve bázy
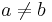
- Pre daný čas t máme teda všade mimo diagonály s(t) a na diagonále 1-3s(t)
- Matica rýchlostí R: pre Jukes-Cantorov model všade mimo diagonály , na diagonále
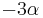
- Pre veľmi malý čas t je S(t) zhruba I-Rt
- Rýchlost alpha je teda pravdepodobnosť zmeny za jednotku casu, ak uvažujeme veľmi krátke časy, resp. derivácia s(t) vzhľadom na t v bode 0
- Riešením diferenciálnych rovníc pre Jukes-Cantorov model dostávame
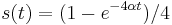
- Matica rýchlostí sa zvykne normalizovať tak, aby na jednotku času pripadla v priemere jedna substitúcia, čo dosiahneme ak
Zložitejšie modely
V praxi sa používajú komplikovanejsie substitučné modely, ktoré majú všeobecnejšiu maticu rýchlostí R
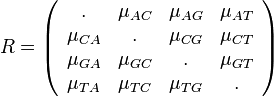
Hodnoty na diagonále matice sa dopočítavajú aby súčet každého riadku bol 0.
- Hodnota
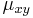 v tejto matici vyjadruje rýchlosť, akou sa určitá báza x mení na inú bázu y.
v tejto matici vyjadruje rýchlosť, akou sa určitá báza x mení na inú bázu y.
- Presnejšie
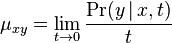 .
.
Kimurov model napr. zachytáva, ze puríny sa častejšie menia na iné puríny (A a G) a pyrimidíny na ine pyrimidíny (C a T).
- Má dva parametre: rýchlosť tranzícií alfa, transverzií beta
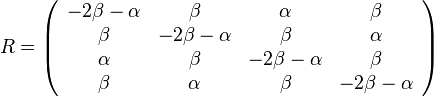
- HKY model (Hasegawa, Kishino & Yano) tiež umožnuje rôzne pravdepodobnosti A, C, G a T v ekvilibriu.
- Ak nastavíme čas v evolučnom modeli na nekonečno, nezáleží na tom, z ktorej bázy sme začali, frekvencia výskytu jednotlivých báz sa ustáli v tzv. ekvilibriu.
- V Jukes-Cantorovom modeli je pravdepodobnosť ľubovoľnej bázy v ekvilibriu 1/4.
- V HKY si zvolime aj frekvencie jednotlivých nukleotidov v ekvilibriu
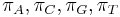 so súčtom 1
so súčtom 1
- Parameter kapa: pomer tranzícií a transverzií (alfa/beta)
- Matica rýchlostí:
-
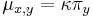 ak mutácia x->y je tranzícia,
ak mutácia x->y je tranzícia,
-
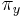 ak mutácia x->y je transverzia
ak mutácia x->y je transverzia
-
- Pre zložité modely nevieme odvodiť explicitný vzorec na výpočet S(t), ako sme mali pri Jukes-Cantorovom modeli
- Ale vo všeobecnosti pre maticu rýchlostí
 dostávame
dostávame 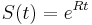 .
.
- Exponenciálna funkcia matice A sa definuje ako
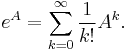
- Ak maticu rychlosti R diagonalizujeme (určite sa dá pre symetrické R)
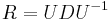 , kde D je diagonálna matica (na jej diagonále budu vlastné hodnoty R), tak
, kde D je diagonálna matica (na jej diagonále budu vlastné hodnoty R), tak 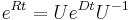 , t.j. exponenciálnu funkciu uplatníme iba na prvky na uhlopriečke matice D.
, t.j. exponenciálnu funkciu uplatníme iba na prvky na uhlopriečke matice D.
- Exponenciálna funkcia matice A sa definuje ako
CB07
Doplnenie prednášok
- Ortológy, paralógy, génové stromy
- Pozitívna selekcia
Fitchov algoritmus

- Parsimony/uspornost
- Vstup: fylogeneticky strom, 1 stlpec zarovnania (jedna baza v kazdom liste stromu)
- Vystup: priradenie baz predkom minimalizujuce pocet substitucii
- Priklad - obr 1
- Uvazujme, co vieme povedat o strome s dvoma susednymi listami vo vacsom strome (oznacenie: obr. 2, listy v1 a v2, hrany do listov e1, e2, ich predok v3, hrana z v3 vyssie e3).
- Ak oba listy maju bazu rovnaku bazu, napr. A, predok v3 v optimalnom rieseni bude urcite mat bazu A
- Dokaz sporom: nech to tak nie je, nech optimalne riesenie ma nejaku inu bazu, napr. C. Vymenme v tomto rieseni toto C za A. Moze nam pribudnut jedna mutacia na hrane e3, ale ubudnu dve na hranach e1 a e2. Tym celkova cena riesenia klesne o 1, takze nebolo optimalne.
- Ak tieto dva listy maju rozne bazy, napr. A a C, tak existuje optimalne riesenie, ktore ma v predkovi v3 bazu A alebo C.
- Dokaz: vezmime optimalne riesenie. Ak ma v3 bazu A alebo C, tvrdenie plati. Ak ma v3 nejaku inu bazu, napr T, mozeme ju vymenit napr. za A, ci mozno pribudne jedna mutacia na e3 ale urcite ubudne mutacia na e1. Teda celkovy pocet mutacii sa nezvysi a nase nove riesenie je stale optimalne. Pozor, vo vseobecnosti nevieme povedat, ci ma v3 mat bazu A alebo C. V niektorych pripadoch su optimalne obe, v niektorych len jedna z nich.
- Fitchov algoritmus 1971
- Kazdemu vrcholu v priradime mnozinu baz M(v)
- M(v) pocitame od listov smerom ku korenu
- Pre list v bude M(v) obsahovat bazu v tomto liste
- Uvazujme vnutorny vrchol v s detmi x a y. Mame uz spocitane M(x) a M(y), chceme M(v)
- Ak M(x) a M(y) maju nejake spolocne bazy, vsetky tieto spolocne bazy dame do M(v), t.j.
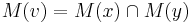
- Ak M(x) a M(y) nemaju spolocne bazy, do M(v) dame vsetky bazy z M(x) aj M(v), t.j.
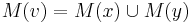
- V tomto pripade pocet mutacii vzrastie o jedna
- Ked mame M(v) spocitane pre vsetky vrcholy, ideme od korena smerom k listom a vyberieme vzdy jednu bazu z M(v).
- Ak sme vybrali pre rodica bazu x a x je v M(v), zvolime x aj pre v, inak zvolime lubovolnu bazu z M(v).
- Priklad algoritmu na obr 3
Gény, evolúcia a komparatívna genomika v UCSC genome browseri (cvičenie pri počítači)
- Zobrazme si gén CLCA4 [17]
- Zapnite si štandardnú sadu track-ov
- Po kliknutí na gén si môžete prečítať o jeho funkcii, po kliknutí na ľavú lištu alebo na názov tracku v zozname na spodku stránky si môžete prečítať viac o tracku a meniť nastavenia
- V tracku RefSeq genes si všimnite, že v tejto databáze má tento gén dve formy zostrihu, jedna z nich sa považuje za nekódujúcu, pretína sa aj s necharakterizovanou nekódujúcou RNA na opačnom vlákne
- Nižšie vidíte track H3K27Ac Mark (Often Found Near Regulatory Elements) on 7 cell lines from ENCODE, kde bola táto histónová modifikácia v okolí génu detegovaná?
- Všimnite si aj track DNase I Hypersensitivity, ktorý zobrazuje otvorený chromatin, prístupný pre viazanie transkripčných faktorov. Všimnite si jeho súvis s H3K27Ac trackom
- Obidva tracky sú súčasťou tracku ENCODE regulation, v ktorom si môžete zapnúť aj ďalšie pod-tracky
- Vsimnime si track Vertebrate Multiz Alignment & Conservation (100 Species)
- v spodnej casti tracku vidime zarovnania s roznymi inymi genomami
- zapnime si v nastavaniach tracku zapnite Element Conservation (phastCons) na full a Conserved Elements na dense
- v tomto tracku vidíme PhyloP, co zobrazuje uroven konzerovanosti danej bazy len na zaklade jedneho stlpca zarovnania a dva vysledky z phyloHMM phastCons, ktory berie do uvahy aj okolite stlpce
- Konkretne cast Conserved elements zobrazuje konkretne useky, ktore su najvac konzervovane
- Ak chceme zistit, kolko percent genomu tieto useky pokryvaju, ideme na modrej liste do casti Tools->Table browser, zvolime group Comparative genomics, track Conservation, table 100 Vert. El, region zvolime genome (v celom genome) a stlacime tlacidlo Summary/statistics, dostaneme nieco taketo:
| item count | 10,350,729 |
| item bases | 162,179,256 (5.32%) |
| item total | 162,179,256 (5.32%) |
| smallest item | 1 |
| average item | 16 |
| biggest item | 3,732 |
| smallest score | 186 |
| average score | 333 |
| biggest score | 1,000 |
- Ak by nas zaujimali iba velmi dlhe "conserved elements", Table browser stlacime tlacidlo Filter a na dalsej obrazovke do policka Free-form query dame chromEnd-chromStart>=1500
- Potom mozeme skusit Summary/Statistics alebo vystup typu Hyperlinks to genome browser a Get output - dostaneme zoznam tychto elementov a kazdy si mozeme jednym klikom pozriet v browseri, napr. taketo
- Pozrime si teraz ten isty gen CLCA4 v starsej verzii genomu hg18 [18]
- V casti Genes and Gene Prediction Tracks zapnite track Pos Sel Genes, ktory obsahuje geny s pozitivnym vyberom (cervenou, pripadne slabsie fialovou a modrou)
- Ked kliknete na cerveny obdlznik pre tento gen, uvidite, v ktorych castiach fylogenetickeho stromu bol detegovany pozitivny vyber
- Po priblizeni do jedneho z exonov [19] vidite dosledky nesynonymnych mutacii
Poznamka: Existuju aj webservery na predikciu pozitivneho vyberu, napriklad tieto dva:
- Selecton, clanok
- Data monkey clanok
- Skusili sme poslat CLCA4 zo 7 cicavcov, subor tu: [20]
Objavenie génu HAR1 pomocou komparatívnej genomiky
- Pollard KS, Salama SR, Lambert N, et al. (September 2006). "An RNA gene expressed during cortical development evolved rapidly in humans". Nature 443 (7108): 167–72. doi:10.1038/nature05113. PMID 16915236. pdf
- Zobrali všetky regióny dĺžky aspoň 100bp s > 96% podobnosťou medzi šimpanzom a myšou/potkanom (35,000)
- Porovnali s ostatnými cicavcami, zistili, ktoré majú veľa mutáci v človeku, ale málo inde (pravdepodobnostný model)
- 49 štatisticky významných regiónov, 96% nekódujúcich oblastiach
- Najvýznamnejší HAR1: 118nt, 18 substitúcii u človeka, očakávali by sme 0.27. Iba 2 zmeny medzi šimpanzom a sliepkou (310 miliónov rokov), ale nebol nájdený v rybách a žabe.
- Nezdá sa byť polymorfný u človeka
- Prekrývajúce sa RNA gény HAR1R a HAR1F
- HAR1F je exprimovaný v neokortexe u 7 a 9 týždenných embrií, neskôr aj v iných častiach mozgu (u človeka aj iných primátov)
- Všetky substitúcie v človeku A/T->C/G, stabilnejšia RNA štruktúra (ale tiež sú blízko k telomére, kde je viacej takýchto mutácii kvôli rekombinácii a biased gene conversion)
Cvičenie pri počítači
- Môžete si pozrieť tento region v browseri: chr20:63102114-63102274 (hg38), pricom ak sa este priblizite, uvidite zarovnanie aj s bazami a mozete vidiet, ze vela zmien je specifickych pre cloveka
CI08
Felsensteinov algoritmus 1981
- Mame dany strom T s dlzkami hran a bazy v listoch (jeden stlpec zarovnania) a model substitucii (zadany napr. maticou rychlosti R). Spocitajme pravdepodobnost, ze z modelu dostaneme prave tuto kombinaciu baz v listoch.
- Oznacenie:
- Nech X_v je premenna reprezentujuca bazu vo vrchole v a nech x_v je konkretna baza v liste v.
- Nech listy su 1..n a vnut. vrcholy n+1..2n-1, pricom koren je 2n-1.
- Nech p_v je rodic vrchola v a nech dlzka hrany z v do rodica je t_v.
- Nech P(a|b,t) je pravdepodobnost, ze b sa zmeni na a za cas t (spocitame z matice R, vid minule cvicenia).
- Napr. v Jukes-Cantorovom modeli ,
- Napr. v Jukes-Cantorovom modeli
- Nech q_a je pravdepodobnost bazy a v koreni (ekvilibrium matice R)
- Napr. v Jukes-Cantorovom modeli q_a = 1/4
- Ak by sme poznali bazy vo vsetkych vrcholoch, mame
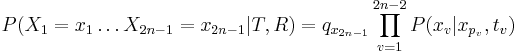
- Chceme pravdepodobnost
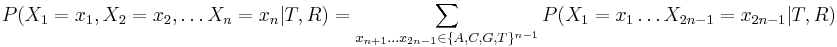
- Pocitat sucet cez exponencialne vela dosadeni hodnot za vnutorne vrcholy je neefektivne, spocitame rychlejsie dynamickym programovanim.
- Nech A[v,a] je pravdepodobnost dat v podstrome s vrcholom v ak X_v=a
- A[v,a] pocitame od listov ku korenu
- V liste A[v,a] = [a=x_v]
- Vo vnut. vrchole s detmi y a z mame
![A[v,a]=\sum _{{b,c}}A[y,b]A[z,c]P(b|a,t_{y})P(c|a,t_{z})](/vyuka/mbi/images/math/e/c/b/ecbada933fd6f145f641b4e2793e58df.png)
- Celkova pravdepodobnost je
![P(X_{1}=x_{1},X_{2}=x_{2},\dots X_{n}=x_{n}|T,R)=\sum _{a}A[r,a]q_{a}](/vyuka/mbi/images/math/3/0/d/30d167737e880c69f2a1cd08a5dfc5a5.png) pre koren r.
pre koren r.
Zlozitost, zlepsenie
- Zlozitost
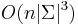
- Pre nebinarne stromy exponencialne
- Zlepsenie
![A[v,a]=(\sum _{{b}}A[y,b]P(b|a,t_{y}))(\sum _{c}A[z,c](c|a,t_{z}))](/vyuka/mbi/images/math/0/2/0/0201f86e4456366fe1c8944f7576e3b5.png)
- Zlozitost
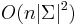 aj pre nebinarne stromy
aj pre nebinarne stromy
Chybajuce data
- Ak v niektorom liste mame neznamu bazu N, nastavime A[v,a]=1
- Podobne sa spracovavaju medzery v zarovnani, aj ked mohli by sme mat aj model explicitne ich modelujuci
Aposteriorna pravdepodobnost (nerobili sme)
- Co ak chceme spocitat pravdepodobnost P(X_v=a|X_1=x_1, X_2=x_2,\dots X_n=x_n,T,R)? Zaujimaju nas teda sekvencie genomov predkov.
- Potrebujeme B[v,a]=pravdpodobnost dat ak podstrom v nahradim listom s bazou a.
- B[v,a] pocitame od korena k listom
- V koreni B[v,a] = q_a
- Vo vrchole v s rodicom u a surodencom x mame
![B[v,a]=\sum _{{b,c}}B[u,b]A[x,c]P(a|b,t_{v})P(c|b,t_{v})](/vyuka/mbi/images/math/0/4/c/04cc0bf38cc6bbf3b4ddf7ce0e346560.png)
- Ziadana pravdepodobnost je
![B[v,a]A[v,a]/P(X_{1}=x_{1},X_{2}=x_{2},\dots X_{n}=x_{n}|T,R)](/vyuka/mbi/images/math/c/c/2/cc2c38f95bb6131cc0a4edfdc7db8bdb.png)
CB08
Na týchto cvičeniach sa budeme venovat dvom statistickym temam suvisiacim s komparativnou genomikou a s analyzou expresie genov. Tieto techniky sa vsak vyuzivaju aj v inych oblastiach a mozete sa s nimi casto stretnut v genomickych clankoch.
Zhlukovanie
- Máme vstupné dáta, väčšinou ako vektory dĺžky n
- Snažíme sa ich rozdeliť do skupín tak, aby dáta v rámci skupiny boli podobné a medzi skupinami rôzne
Využitie:
- hľadanie génov s podobným profilom expresie
- hľadanie skupín pacientov s podobným profilom expresie génov (objavovanie podtypov nejakej choroby)
- hľadanie rodín podobných proteínov
- automatická segmentácia obrázkov (napríklad rozlíšiť jednotlivé políčka microarray alebo gelu od pozadia)
Na prednáške sme videli hierarchické zhlukovanie, ktoré z dát vytvorilo strom. Teraz si ukážeme zhlukovanie, ktoré sa snaží dáta rozdeliť na k skupín, kde k je vopred daný parameter.
K-Means


- pozri tiež prezentáciu pdf
- Vstup: n-rozmerné vektory
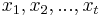 a počet zhlukov k
a počet zhlukov k
- Výstup: Rozdelenie vektorov do k zhlukov takéto:
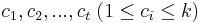 - priradenie vektoru k zhluku
- priradenie vektoru k zhluku
- n-rozmerné vektory
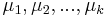 - centrá každého zhluku
- centrá každého zhluku
- Úloha: minimalizovať súčet štvorcov vzdialeností od každého vektoru k centru jeho zhluku:
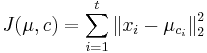
-
 je druhá mocnina vzdialenosti vektora xi od centra jeho zhluku
je druhá mocnina vzdialenosti vektora xi od centra jeho zhluku
-
Algoritmus
Heuristika, ktorá nenájde vždy najlepšie zhlukovanie. Začne z nejakého zhlukovania a postupne ho zlepšuje. Pozri aj clanok na Wikipedii
- inicializácia: náhodne vyber k centier
- opakuj kým sa niečo mení:
- priraď každý bod najbližšiemu centru:
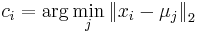
- vypočítaj nové centroidy:
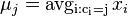 (spriemerujeme všetky body v jednom zhluku)
(spriemerujeme všetky body v jednom zhluku)
- priraď každý bod najbližšiemu centru:
Nadreprezentacia, obohatenie (enrichment)
- Mnohe celogenomove analyzy nam daju zoznam genov, ktore sa v nejakom ukazovateli vyrazne lisia od priemeru.
- Napriklad geny s pozitivnym vyberom v komparativnej genomike, geny vyrazne nadexprimovane alebo podexprimovane v microrarray experimentoch, geny regulovane urcitym transkripcnym faktorom a pod.
- Niektore z nich budu preskumanejsie (znama funkcia a pod.), niektore mozu mat nejake udaje o funkcii prenesene z homologov a dalsie mozu byt uplne nezname
- Co s takym zoznamom "zaujimavych genov"?
- moznost 1: vybrat si z neho niekolko malo zaujimavych kandidatov a preskumat ich podrobnejsie (experimentalne alebo informaticky)
- moznost 2: zistit, ci tato cela skupina je obohatena o geny urcitych skupin
- napr. v pripade pozitivneho vyberu nam casto vychadzaju geny suvisiace s imunitou, lebo su pod velkym evolucnym tlakom od patogenov
- takato analyza nam teda da informaciu o suvislostiach medzi roznymi procesmi
- Priklad (Kosiol et al)
- 16529 genov celkovo, 70 genov v GO kategorii innate immune response (0.4% zo vsetkych genov)
- 400 genov s pozivnym vyberom, mame 8 genov s innate immune response (2% zo vsetky genov s poz. vyb.)
- Celkovy pocet genov n, imunitnych ni, pozitivny vyber np, imunitnych s poz. vyb. nip.
- Kontingencna tabulka
| Pozitivny vyber | Bez poz. vyberu | Sucet | |
|---|---|---|---|
| Imunitne | 8 (nip) | 62 | 70 (ni) |
| Ostatne | 392 | 16067 | 16459 |
| Sucet | 400 (np) | 16129 | 16529 (n) |
- Nulova hypoteza: geny v nasom zozname boli nahodne vybrane z celeho genomu, t.j. ak v celom genome je frekvencia imunitnych genov ni/n (cca 0.4%), vo vzorke velkosti np (geny s pozitivnym vyberom) ocakavame cca np * (ni / n) imunitnych genov.
- aj v nulovej hypoteze vsak vzorka velkosti ni cisto nahodou moze obsahovat viac alebo menej takych genov.
- presnejsie mame urnu so ni (70) bielymi a n-ni (16459) ciernymi gulickami, vytiahneme nahodne np (400) guliciek, kolko bude medzi nimi bielych, nazvime tuto nahodnu premennu Xip
- v nasom priklade by sme ocakavali 1.7 genu s innate immune response, ale mame 8 (4.7xviac)
- Rozdelenie pravdepodobnosti Xip je hypergeometricke, t.j.
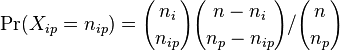
- Aka je pravdepodobnost, ze v nulovej hypoteze bude Xip tolko, kolko sme namerali alebo viac? (Chvost rozdelenia). V nasom pripade p-value 2.8e-4.
- Hypergeometric or Fisher's exact test, pripadne ich aproximacie pre velke hodnoty v tabulke (chi^2 test) zisti, ci sa nasa tabulka velmi lisi od toho, co by sme ocakavali v nulovej hypoteze
- Suvisiace clanky
- Rivals I, Personnaz L, Taing L, Potier MC (February 2007). "Enrichment or depletion of a GO category within a class of genes: which test?". Bioinformatics (Oxford, England) 23 (4): 401–7. doi:10.1093/bioinformatics/btl633. PMID 17182697.
- Huang da W, Sherman BT, Lempicki RA (January 2009). "Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists". Nucleic Acids Research 37 (1): 1–13. doi:10.1093/nar/gkn923. PMID 19033363.
- Existuju web servery, napr. GOrilla pre ludske geny: http://cbl-gorilla.cs.technion.ac.il/, DAVID (http://david.niaid.nih.gov), g:Profiler http://biit.cs.ut.ee/gprofiler/
- Treba dat pozor, ci pocitaju to co chceme
- Kod v statistickom systeme R na pocitanie hypergeometrickeho rozdelenia
> dhyper(0:70, 70, 16529-70, 400); [1] 1.793421e-01 3.126761e-01 2.679872e-01 1.505169e-01 6.231088e-02 [6] 2.027586e-02 5.400796e-03 1.210955e-03 2.332580e-04 3.920215e-05 [11] 5.818723e-06 7.702558e-07 9.166688e-08 9.873221e-09 9.678760e-10 [16] 8.677204e-11 7.143849e-12 5.420388e-13 3.802134e-14 2.472342e-15 [21] 1.493876e-16 8.405488e-18 4.412274e-19 2.164351e-20 9.935473e-22 [26] 4.273662e-23 1.724446e-24 6.533742e-26 2.326517e-27 7.791092e-29 [31] 2.455307e-30 7.285339e-32 2.036140e-33 5.361856e-35 1.330660e-36 [36] 3.112566e-38 6.862558e-40 1.426089e-41 2.792792e-43 5.153006e-45 [41] 8.955105e-47 1.465159e-48 2.255667e-50 3.265636e-52 4.442631e-54 [46] 5.674366e-56 6.797781e-58 7.629501e-60 8.012033e-62 7.860866e-64 [51] 7.193798e-66 6.129013e-68 4.851139e-70 3.558526e-72 2.412561e-74 [56] 1.506983e-76 8.641725e-79 4.530590e-81 2.161126e-83 9.326620e-86 [61] 3.617279e-88 1.250737e-90 3.817900e-93 1.016417e-95 2.323667e-98 [66] 4.469699e-101 7.034762e-104 8.698702e-107 7.924236e-110 4.728201e-113 [71] 1.386176e-116 phyper(7, 70, 16529-70, 400, lower.tail=FALSE); # pr pocet bielych>7 (t.j. >=8) ak taham 400 z vreca so 70 bielymi a 16529-70 ciernymi # sucet cisiel z tabulky od 2.332580e-04 az po koniec d = dhyper(0:15, 70, 16529-70, 400); plot(0:15,d)
Multiple testing correction
- V mnohych situaciach robime vela testov toho isteho typu, kazdy ma urcitu p-value
- Napr. testujeme 1000 genov v genome na pozitivny vyber, zvolime tie, kde p-value <= 0.05
- Alebo testujeme obohatenie 1000 funkcnych kategorii v nejakej vzorke genov, zvolime tie, kde p-value <= 0.05
- Problem: ak kazda z 1000 kategorii ma 5% sancu tam byt len nahodou, ocakavali by sme 50 cisto nahodnych pozitivnych vysledkov. Ak sme napr. nasli 100 pozitivnych vysledkov (obohatenych kategorii), cca polovica z nich je zle
- Preto potrebujeme pri velkom mnozstve testov umelo znizit prah na p-value tak, aby nahodny sum netvoril velke percento nasich vysledkov
- Toto sa vola multiple testing correction, je viac technik, napr. FDR (false discovery rate)
Prakticke cvicenie pri pocitaci
Data o expresii ludskych genov v roznych tkanivach a podobne v UCSC genome browseri
- Chodte na genome browser http://genome-euro.ucsc.edu/
- Zvolte Tools->Gene Sorter, sort by zvolme Expression (GNF Atlas 2), a do okienka search zadajme identifikator genu PTPRZ1
- Dostane tabulku genov s podobnym profilom expresie ako PTPRZ1 (červená je vysoká expresia, zelená nízka)
- Zoznam tychto genov v textovom formate najdete tu
- http://biit.cs.ut.ee/gprofiler/ mena genov skopirujme do policka Query, stlacte g:Profile!
- Vo vyslednej tabulke je kazdy riadok jedna funkcna kategoria, v ktorej su geny s tymto profilom expresie nadreprezentovane, kazdy stlpec jeden gen.
- Co by sme na zaklade nadreprezentovanych kategorii usudzovali o gene PTPRZ1?
- Najdite tento gen v Uniprote (http://www.uniprot.org/), potvrdzuje nase domnienky?
- Vratme sa do genome browsera, najdime si PTPRZ1 gen v genome
- V browseri su rozne tracky tykajuce sa expresie, napr. GNF Atlas 2. Precitajte si, co je v tomto tracku zobrazene, zapnite si ho a pozrite si expresiu okolitych genov okolo PTPRZ1
- Kliknite na gen v tracku UCSC known genes. V tabulke uvidite zase prehlad expresie v roznych tkanivach (podla GNF Atlasu)
NCBI Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo/
- Databaza gene expression dat na NCBI
- Do Search okienka zadajme GDS2925
- Mali by sme dostat dataset Various weak organic acids effect on anaerobic yeast chemostat cultures
- Mozeme si pozriet zakladne udaje, napr. citation, platform
- Link "Expression profiles" nam zobrazi grafy pre rozne geny
- Pri kazdom profile mozeme kliknut na profile neighbors, aby sme videli geny s podobnym profilom
- Data analysis tools, cast Cluster heatmaps, K-means, skuste rozne pocty clustrov
CI09
Hladanie motivov zadefinovanych pravdepodobnostnou maticou
- Mame danych n sekvencii
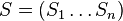 , kazda dlzky m, dlzku motivu L, nulova hypoteza q (frekvencie nukleotidov v genome)
, kazda dlzky m, dlzku motivu L, nulova hypoteza q (frekvencie nukleotidov v genome)
- Hladame motiv vo forme pravdepodobnostneho profilu dlzky L a jeho vyskyt v kazdej sekvencii
- Nech
![W[a,i]](/vyuka/mbi/images/math/0/3/2/032862e9742350e6fd5f134812164f21.png) je pravdepodobnost, ze na pozicii i motivu bude baza a, W cela matica
je pravdepodobnost, ze na pozicii i motivu bude baza a, W cela matica
-
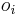 je pozicia vyskytu v sekvencii
je pozicia vyskytu v sekvencii 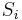 ,
, 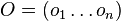 su vsetky vyskyty
su vsetky vyskyty
-
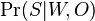 je jednoduchy sucin, kde pre pozicie v oknach pouzijeme pravdepodobnosti z W, pre pozicie mimo okna pouzijeme q
je jednoduchy sucin, kde pre pozicie v oknach pouzijeme pravdepodobnosti z W, pre pozicie mimo okna pouzijeme q
-
- Hladame W a O, ktore maximalizuju tuto vierohodnost Pr(S|W,O)
- Nepozname efektivny algoritmus, ktory by vedel vzdy najst maximum
- Dali by sa skusat vsetky moznosti O, pre dane O je najlepsie W frekvencie z dat
- Naopak ak pozname W, vieme najst najlepsie O
- v kazdej sekvencii i skusame vsetky pozicie a zvolime tu, ktora ma najvyssiu hodnotu
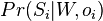
- v kazdej sekvencii i skusame vsetky pozicie
![\Pr(S_{i}|W,o_{i})=\prod _{{j=1}}^{{L}}W[S_{i}[j+o_{i}-1],j]\prod _{{j=1}}^{{o_{i}-1}}q[S_{i}[j]]\prod _{{j=o_{i}+L}}^{m}q[S_{i}[j]]](/vyuka/mbi/images/math/5/f/a/5fadd02fa29abf436fdd8a9d2f225bfe.png)
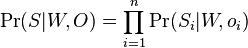
![[14]](http://genome-euro.ucsc.edu/images/phylo/hg38_100way.png){kind=link}
EM algoritmus
- iterativne zlepsuje W, pricom berie vsetky O vahovane podla ich pravdepodobnosti vzhladom na W z minuleho kola
- Videli sme na prednaske, tu je trochu prepisany:
- Inicializácia: priraď každej pozícii j v sekvencii nejaké skóre
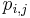
- Iteruj:
- Spočítaj W zo všetkých možných výskytov v
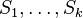 váhovaných podľa
váhovaných podľa
- Prepočítaj všetky skóre tak, aby zodpovedali pomerom pravdepodobností výskytu W na pozícii j v , t.j. je umerne
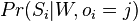 , pricom hodnoty normalizujeme tak, aby sucet v riadku bol 1
, pricom hodnoty normalizujeme tak, aby sucet v riadku bol 1
- Spočítaj W zo všetkých možných výskytov v
Gibbsovo vzorkovanie (Gibbs sampling)
- Inicializácia: Vezmi náhodné pozície výskytov O
- Iteruj:
- Spočítaj W z výskytov O
- Vyber náhodne jednu sekvenciu
- Pre každú možnú pozíciu j v spočítaj skóre (ako v EM) výskytu W na tejto pozícii
- Zvoľ náhodne s váhovaním podľa
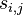
- Takto dostavame postupnost vzoriek
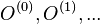 .
.
- Za sebou iduce vzorky sa podobaju (lisia sa len v jednej zlozke ) nie su teda nezavisle
- Pre kazdu vzorku
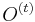 najdeme najlepsie
najdeme najlepsie 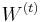 a spocitame vierohodnost
a spocitame vierohodnost 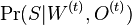 . Nakoniec vyberieme O a W, kde bola vierohodnost najvyssia.
. Nakoniec vyberieme O a W, kde bola vierohodnost najvyssia.
- Tento algoritmus (s malymi obmenami) bol pouzity v clanku Lawrence, Charles E., et al. (1993) "Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment." Science.
- V clanku v kazdej iteracii maticu W rataju zo vsetkych sekvencii okrem
- Obcas robia krok, kde nahodne skusaju posunut vsetky vyskyty o jedna dolava alebo doprava
- Tento algoritmus nie je uplne matematicky korektne Gibbsovo vzorkovanie (nema ani poradne zadefinovane rozdelenie, z ktoreho vzorkuje). Na spodku stranky pre informaciu uvadzame algoritmus Gibbsovho vzorkovanie pre hladanie motivov z ineho clanku.
- V clanku v kazdej iteracii maticu W rataju zo vsetkych sekvencii okrem
Vzorkovanie z pravdepodobnostneho modelu vo vseobecnosti
- majme pravdepodobnostny model, kde D su nejake pozorovane data a X nezname nahodne premenne (napr pre nas D su sekvencie S a X su vyskyty O, pripadne aj matica W)
- mozeme hladat X pre ktore je vierohodnost Pr(D|X) najvyssia
- alebo mozeme nahodne vzorkovat rozne X z Pr(X|D)
Pouzitie vzoriek
- spomedzi ziskanych vzoriek zvolime tu, pre ktoru je vierohodnost Pr(D|X) najvacsia (iny pristup k maximalizovaniu vierohodnosti)
- ale vzorky nam daju aj informaciu o tom, aka je velka neurcitost v odhade X
- mozeme odhadovat stredne hodnoty a odchylky roznych velicin
- napr. pri hladani motivov mozeme sledovat ako casto je ktora pozicia vyskytom motivu
- generovat nezavisle vzorky z Pr(X|D) moze byt tazke
- metoda Markov chain Monte Carlo (MCMC) generuje postupnost zavislych vzoriek
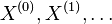 , konverguje v limite k cielovej distribucii Pr(X|D)
, konverguje v limite k cielovej distribucii Pr(X|D)
- Gibbsovo vzorkovanie je specialnym pripadom MCMC
Markovove reťazce
- Markovov reťazec je postupnosť náhodných premenných
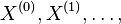 taká, že
taká, že 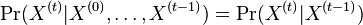 , t.j. hodnota v čase závisí len od hodnoty v čase
, t.j. hodnota v čase závisí len od hodnoty v čase 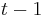 a nie ďalších predchádzajúcich hodnôt.
a nie ďalších predchádzajúcich hodnôt.
- Nás budú zaujímať homogénne Markovove reťazce, v ktorých
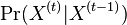 nezávisí od .
nezávisí od .
- Tiez nas zaujimaju len retazce v ktorych nahodne premenne
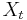 nadobudaju hodnoty z konecnej mnoziny (mozne hodnoty
nadobudaju hodnoty z konecnej mnoziny (mozne hodnoty 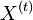 nazyvame stavy)
nazyvame stavy)
- Napriklad stavy A,C,G,T
- V Gibbsovom vzorkovani pre motivy je stav konfiguracia premennych O (t.j. mame (m-L+1)^n stavov)
- Vzorka v kroku t zavisi od vzorky v kroku t-1 (a lisi sa len v hodnote jedneho o_i)
Matica
- Pravdepodobnosti prechodu medzi stavmi za jeden krok mozeme vyjadrit maticou pravdepodobnosti P, ktorej prvok
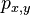 oznacuje pravdepodobnost prechodu zo stavu x do stavu y
oznacuje pravdepodobnost prechodu zo stavu x do stavu y 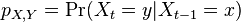
- Sucet kazdeho riadku je 1, cisla nezaporne
- Ako
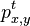 budeme oznacovat
budeme oznacovat 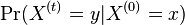 , tieto hodnoty dostaneme umocnenim matice P na t
, tieto hodnoty dostaneme umocnenim matice P na t
Stacionarne rozdelenie
- Rozdelenie na mnozine stavov sa nazyva stacionarne pre Markovov retazec
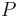 , ak pre kazde j plati
, ak pre kazde j plati 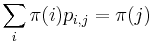 (alebo v maticovej notacii
(alebo v maticovej notacii 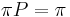 )
)
- Ak matica P splna urcite podmienky (je ergodicka), existuje pre nu prave jedno stacionarne rozdelenie . Navyse pre kazde x a y plati
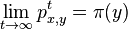
Priklady Markovovskych retazcov v bioinformatike
- V HMM stavy tvoria Markovov retazec
- Ine varianty: nekonecne stavove priestory (zlozitejsia teoria), spojity cas (videli sme pri evolucnych modeloch), retazce vyssieho radu, kde urcujeme
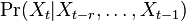 a pod.
a pod.
- Pouzitie v bioinformatike: charakterizacia nahodnych sekvencii (nulova hypoteza), pre DNA sa pouzivaju rady az do 5, lepsie ako nezavisle premenne
Ergodické Markovove reťazce
- Vravime ze matica je ergodicka, ak
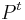 pre nejake t>0 ma vsetky polozky nenulove
pre nejake t>0 ma vsetky polozky nenulove
- Priklady neergodickych matic
1 0 0.5 0.5 0 1 0.5 0.5 0 1 0 1 1 0 1 0 nesuvisla slabo suvisla periodicka ergodicka
- V HMM stavy tvoria Markovov retazec; hladanie genov ergodicky stavovy priestor, profilove HMM nie
Markov chain Monte Carlo MCMC
- Chceme generovať náhodné vzorky z nejakeho cieloveho rozdelenia , ale toto rozdelenie je prilis zlozite.
- Zostavime ergodicky Markovov retazec, ktoreho stacionarne rozdelenie je rozdelenie , tak aby sme efektivne vedeli vzorkovat ak vieme
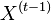 .
.
- Ak zacneme z lubovolneho bodu
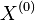 , po urcitom case t rozdelenie priblizne
, po urcitom case t rozdelenie priblizne
- Ale za sebou iduce vzorky nie su nezavisle!
- Vieme vsak odhadovat ocakavane hodnoty roznych velicin
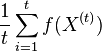 konverguje k
konverguje k ![E_{\pi }[f(X)]](/vyuka/mbi/images/math/a/a/4/aa4ecd5f5f91f0aaf04047c7435e1922.png)
Gibbsovo vzorkovanie
- Cielove rozdelenie
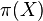 je cez vektory dlzky n
je cez vektory dlzky n 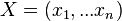
- V kazdom kroku vzorkujeme jednu zlozku vektora
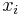 z podmienenej pravdepodobnosti
z podmienenej pravdepodobnosti 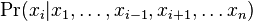
- Ostatne hodnoty nechame rovnake ako v predchadzajucom kroku
- Hodnotu zvolime nahodne alebo periodicky striedame
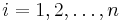
Dokaz spravnosti Gibbsovho vzorkovania
- Pozor! Gibbsovo vzorkovanie nie je vzdy ergodicke, ak niektore kombinacie hodnot maju nulovu pravdepodobnost!
- Treba dokazat, ze ak je ergodicky, tak ma ako stacionarnu distribuciu nase zvolene
- Definicia: Vravime, ze matice P a rozdelenie splnaju detailed balance, ak pre kazde stavy (dva vektory hodnot) x a y mame
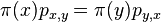
- Lema: ak pre nejaky retazec P a nejake rozdelenie plati detailed balance, je stacionarna distribucia pre P
- Dokaz:
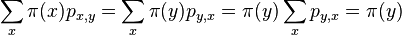
- Dokaz:
- Lema: pre retazec Gibbsovo vzorkovania plati detailed balance vzhladom na cielove rozdelnie
- Dokaz: uvazujme dva za sebou iduce vektory hodnot x a y, ktore sa lisia v i-tej suradnici. Nech
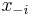 su hodnoty vsetkych ostatnych premennych okrem
su hodnoty vsetkych ostatnych premennych okrem
-
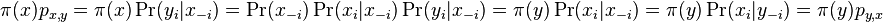
- Dokaz: uvazujme dva za sebou iduce vektory hodnot x a y, ktore sa lisia v i-tej suradnici. Nech
Poriadnejsie Gibbsovo vzorkovanie pre motivy
Uvedene pre zaujimavost - podla clanku Siddharthan R, Siggia ED, van Nimwegen E (December 2005). "PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny". PLoS Comput. Biol. 1 (7): e67. doi:10.1371/journal.pcbi.0010067. PMID 16477324.
Pravdepodobnostny model
- Rozsirime model, aby aj O a W boli nahodne premenne, takze mame rozdelenie Pr(S,W,O)
- Potom chceme vzorkovat z Pr(O|S) (marginalizujeme cez vsetky hodnoty W)
- Vygeneruje sa nahodne matica pravdepodobnosti W (napr z roznomernej distribucie cez vsetky matice)
- V kazdej sekvencii i sa zvoli okno dlzky L (rovnomerne z m-L+1 moznosti)
- V okne sa generuje sekvencia podla profilu W a mimo okna sa generuje sekvencia z nulovej hypotezy (ako predtym)
Gibbsovo vzorkovanie
- Mame dane S, vzorkujeme O (
 ) (ak treba, z mozeme zostavit maticu )
) (ak treba, z mozeme zostavit maticu )
- zacni s nahodnymi oknami
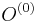
- v kroku t+1 zvol jednu sekvenciu i a pre vsetky pozicie
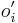 spocitaj
spocitaj 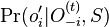 (kde
(kde  , t.j. všetky pozície výskytov okrem i-tej).
, t.j. všetky pozície výskytov okrem i-tej).
- nahodne zvol jedno umerne k tymto pravdepodobnostiam
-
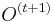 dostaneme z vymenou pozicie v sekvencii i za prave zvolenu
dostaneme z vymenou pozicie v sekvencii i za prave zvolenu
- opakuj vela krat
- zacni s nahodnymi oknami
- Konverguje k cielovemu rozdeleniu
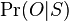 , ale vzorky nie su nezavisle
, ale vzorky nie su nezavisle
- Dalsie mozne kroky vo vzorkovani: posun vsetky okna o konstantu vlavo alebo vpravo
- Dalsie moznosti rozsirenia modelu/algoritmu: pridaj rozdelenie cez L a nahodne zvacsuj/zmensuj L, dovol vynechat motiv v niektorych sekvenciach, hladaj viac motivov naraz,...
Ako spocitat 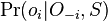 ?
?
- nezaujimaju nas normalizacne konstanty, lahko znormalizujeme scitanim cez vsetky
-
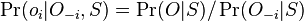 , ale menovatel konstanta
, ale menovatel konstanta
-
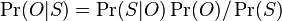 , kde
, kde 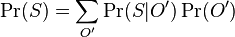
- Menovatel nas nezaujima (normalizacna konstanta)
-
 je tiez konstanta (rovnomerne rozdelenie pozicii okien)
je tiez konstanta (rovnomerne rozdelenie pozicii okien)
- Teda mame je umerne
- Lahko vieme spocitat , potrebujeme "zrusit" W, da sa spocitat vzorec...
- Skusame vsetky mozne hodnoty , pocitame pravdepodobnost , vzorkujeme umerne k tomu
Dalsie detaily vypoctu :
- Nech
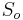 su len sekvencie v oknach a
su len sekvencie v oknach a 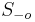 mimo okien. Mame
mimo okien. Mame 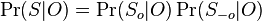
-
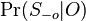 lahko spocitame (nezavisi od W)
lahko spocitame (nezavisi od W)
-
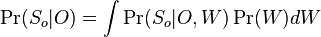 kde integral ide cez hodnoty, kde
kde integral ide cez hodnoty, kde 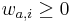 a
a 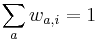
-
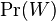 je konstanta (rovnomerne rozdelenie; nejde o pravdepodobnost ale hustotu),
je konstanta (rovnomerne rozdelenie; nejde o pravdepodobnost ale hustotu), 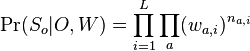 , kde
, kde 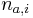 je pocet vyskytov bazy a na pozicii i v oknach
je pocet vyskytov bazy a na pozicii i v oknach 
-
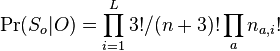 (bez dokazu)
(bez dokazu)
CB09
Bezkontextové gramatiky
- Na modelovanie struktury RNA sa pouzivaju stochasticke bezkontextove gramatiky (bude na dalsej prednaske)
- My si teraz ukazeme bezkontextove gramatiky, ktore nemaju pravdepodobnosti
- Zaviedol Noam Chomsky v lingvistike 50-te roky 20. storocia, tiez dolezite v informatike
Gramatika
- Priklad: S->aSb, S->epsilon (piseme aj skratene S->aSb|epsilon)
- Dva typy symbolov: terminaly (male pismena), neterminaly (velke pismena)
- Pravidla prepisujuce neterminal na retazec terminalov a neterminalov (moze byt aj prazdny retazec, ktory oznacujeme epsilon)
- Neterminal S je "startovaci"
Pouzitie gramatiky na generovanie retazcov
- Zacneme so startovacim neterminalom S
- V kazdom kroku prepiseme najlavejsi neterminal podla niektoreho pravidla
- Skoncime, ked nezostanu ziadne neterminaly
- Priklad: S->aSb->aaSbb->aaaSbbb->epsilon
- Ake vsetky slova vie tato gramatika generovat?
- V tvare aa...abb...b s rovnakym poctom acok a bciek (informatici pisu
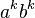 )
)
- V tvare aa...abb...b s rovnakym poctom acok a bciek (informatici pisu
Cvicenia
- Zostavte gramatiku na slova typu aa..abb..b kde acok je rovnako alebo viac ako bcok,
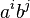 pre
pre 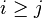
- S->aSb|aS|epsilon
- Zostavte gramatiku pre slova toho isteho typu, kde acok je viac ako bcok, t.j. i>j
- S->aSb|aT T->aT|epsilon (alebo S->aSb|aS|a)
- Zostavte gramatiku pre dobre uzatvorkovane vyrazy zo zatvoriek (,),[,]. Napr. [()()([])] je dobre uzatvorkovany, ale [(]) nie je.
- S->SS|(S)|[S]|epsilon
- priklad odvedenia v tejto gramatike: S->[S]->[SS]->[SSS]->[(S)SS]->[()SS]->[()(S)S]->[()()S]->[()()(S)]->[()()([S])]->[()()([])]
Parsovanie retazca pomocou gramatiky: urcit, ako mohol byt retazec vygenerovany pomocou pravidiel
- Gramatika pre dobre uzatvrokovane vyrazy nam pomoze urcit, ktora zatvorka patri ku ktorej: tie, ktore boli vygenerovane v jednom kroku
Dalsie cvicenia
- Zostavte gramatiku na DNA palindromy, t.j. sekvencie, ktore zozadu po skomplementovani baz daju to iste, ako napr. GATC
- S->gSc|cSg|aSt|tSa|epsilon
- Tazsi priklad: Zostavte gramatiku na slova s rovnakym poctom acok a bcok v lubovolnom poradi
- S->epsilon|aSbS|bSaS
- ako bude generovat aababbba?
- preco vie vygenerovat vsetky take retazce?
Microarray dáta, nadreprezentácia (cvičenie pri počítači)
Data o expresii ludskych genov v roznych tkanivach a podobne v UCSC genome browseri
- Chodte na genome browser http://genome-euro.ucsc.edu/
- Zvolte Tools->Gene Sorter, sort by nechajme Expression (GTEx), a do okienka search zadajme identifikator genu PTPRZ1
- Dostane tabulku genov s podobnym profilom expresie ako PTPRZ1 (červená je vysoká expresia, zelená nízka)
- Zoznam tychto genov v textovom formate najdete tu
- http://biit.cs.ut.ee/gprofiler/ mena genov skopirujme do policka Query, stlacte g:Profile!
- Ak by výpočet dlho trval, nájdete ho aj tu
- Vo výslednej tabuľke je každý riadok jedna funkcna kategoria, v ktorej su geny s tymto profilom expresie nadreprezentovane, kazdy stlpec jeden gen.
- V spodnej casti tabuly su aj asociacie k chorobam a k transkripcnym faktorom, ktore by mohli prislusne geny regulovat
- Co by sme na zaklade nadreprezentovanych kategorii usudzovali o gene PTPRZ1?
- Najdite tento gen v Uniprote (http://www.uniprot.org/), potvrdzuje nase domnienky?
- Vratme sa do genome browsera, najdime si PTPRZ1 gen v genome
- V browseri su rozne tracky tykajuce sa expresie, napr. GTEx. Precitajte si, co je v tomto tracku zobrazene, zapnite si ho a pozrite si expresiu okolitych genov okolo PTPRZ1
- Kliknite na gen v tracku UCSC known genes. V tabulke uvidite zase prehlad expresie v roznych tkanivach (podla GTEx)
NCBI Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo/
- Databaza gene expression dat na NCBI
- Do Search okienka zadajme GDS2925
- Mali by sme dostat dataset Various weak organic acids effect on anaerobic yeast chemostat cultures
- Mozeme si pozriet zakladne udaje, napr. citation, platform
- Link "Expression profiles" nam zobrazi grafy pre rozne geny
- Pri kazdom profile mozeme kliknut na profile neighbors, aby sme videli geny s podobnym profilom
- Data analysis tools, cast Cluster heatmaps, K-means, skuste rozne pocty clustrov
Proteíny
- Toto cvičenie je z časti inšpirované stránkou [23]
- Pozrieme sa na enzým Bis(5'-adenosyl)-triphosphatase
- Nájdime ho na stránke http://www.uniprot.org/ pod názvom FHIT_HUMAN
- O mnohých údajoch na stránke sme sa rozprávali na prednáške (GO kategórie, domény, sekundárna a 3D štruktúra)
- na veľa miestach na stránke je uvedené aj odkiaľ jednotlivé údaje pochádzajú
- Všimnime si Pfam doménu a pozrime si jej stránku [24], do akej super-rodiny (klanu) patrí?
- Tento enzým je vzdialene podobný na enzým galactose-1-phosphate uridylyltransferase (GALT/GAL7)
- Jeho domény patria v databáze Pfam do toho istého klanu, ako doména proteínu FHIT
- Skúsme nájsť túto podobnosť v BLASTe: http://blast.ncbi.nlm.nih.gov/ v časti proteíny, zvoľme databázu Swissport, ako Query zadajme Accesion nášho proteínu P49789, spustie program PSI-BLAST
- V prvom kole PSI-BLAST spúšťa bežný BLASTP
- GAL gén (konkrétne GAL7_HAEIN, accession P31764) sa nachádza medzi výsledkami, ale má príliš vysokú E-value)
- Spustíme teraz druhú iteráciu PSI-BLAST, ktorá zostaví profil z proteínov s nízkou E-value v prvej iterácii
- Aká je E-value nájdeného zarovnania?
CI10
Protein threading
Prakticke programy na NP tazke problemy
- Obcas chceme najst optimalne riesenie nejakeho NP-tazkeho problemu
- Jedna moznost je previest na iny NP tazky problem, pre ktory existuju pomerne dobre prakticke programy, napriklad integer linear programming (ILP)
- najdu optimalne riesenie, mnohe instancie zrataju v rozumnom case, ale mozu bezat aj velmi dlho
- CPLEX [25] a Gurobi [26] komercne baliky na ILP, akademicka licencia zadarmo
- SCIP [27] nekomercny program pre ILP
- SYMPHONY v projekte COIN-OR [28]
- Minisat [29] open source SAT solver
- Concorde TSP solver [30] - riesi problem obchodneho cestujuceho so symetrickymi vzdialenostami, zadarmo na akademicke ucely
- Pre zaujimavost: TSP art [31]
ILP
Linearny program:
- Mame realne premenne x_1...x_n, minimalizujeme nejaku ich linearnu kombinaciu
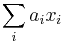 kde a_i su dane vahy.
kde a_i su dane vahy.
- Mame tiez niekolko podmienok v tvare linearnych rovnosti alebo nerovnosti, napr.
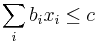
- Hladame teda hodnoty premennych, ktore minimalizuju cielovu sumu, ale pre ktore platia vsetky podmienky
- Da sa riesit v polynomialnom case
Integer linear program
- Program, v ktorom vsetky/vybrane premenne musia mat celociselne hodnoty, alebo dokonca povolime oba hodnoty 0 a 1.
- NP uplny problem
Ako zapisat (NP-tazke) problemy ako ILP
Knapsack
- Problem: mame dane predmety s hmotnostami w_1..w_n a cenami c_1..c_n, ktore z nich vybrat, aby celkova hmotnost bola najviac T a cena bola co najvyssia?
- Pouzijeme binarne premenne x_1..x_n, kde x_i = 1 prave vtedy ked sme zobrali i-ty predmet.
- Chceme maximalizovat
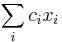
- za podmienky ze
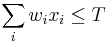
Set cover:
- Mame n mnozin S_1...S_n nad mnozinou {1...m}. Chceme vybrat co najmensi pocet zo vstupnych mnozin tak, aby ich zjednotenie bola cela mnozina {1..m}
- Binarne premenne x_i=1 ak vyberieme i-tu mnozinu
- Chceme minimalizovat
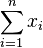
- za podmienky, ze pre kazde j z {1..m} plati
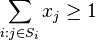
Protein threading
- Ciel: protein A ma znamu sekvenciu aj strukturu, protein B iba sekvenciu. Chceme zarovnat proteiny A a B, pricom budeme brat do uvahy znamu strukturu, t.j. ak su dve amino kyseliny blizko v A tak ich ekvivalenty v B by mali byt "kompatibilne".
- Tento problem chceme riesit tak, ze v strukture A urcime nejake jadra, ktore by v evolucii mali zostat zachovane bez inzercii a delecii a v rovnakom poradi. Tieto jadra su oddelene sluckami, ktorych dlzka sa moze lubovolne menit a ktorych zarovnania nebudeme skorovat.
- Formulacia problemu: Mame danu sekvenciu B=b1..bn, dlzky m jadier c_1...c_m a skorovacie tabulky E_ij, ktora vyjadruje, ako dobre bj..b_{j+c_i-1} sedi do sekvencie jadra i a E_ijkl ktora vyjadruje, ako dobre by jadra i a k interagovali, keby mali sekvencie zacinajuce v B na poziciach j a l. Uloha je zvolit polohy jadier x_1<x_2<...<x_m tak, aby sa ziadne dve jadra neprekryvali a aby sme dosiahli najvyssie skore.
- Poznamka: nevraveli sme, ako konkretne zvolit jadra a skorovacie tabulky, co je modelovaci, nie algoritmicky problem (mozeme skusit napr. nejake pravdepodobnostne modely)
Protein threading ako ILP
- Premenne v programe:
- x_ij=1 ak je zaciatok i-teho jadra zarovnane s b_j
- y_ijkl=1 ak je zaciatok i-teho jadra na b_j a zaciatok k-teho na b_l (i<k, j<l)
- Chceme maximalizovat
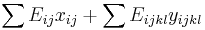
- Podmienky:
-
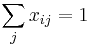 pre kazde i
pre kazde i
-
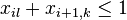 pre vsetky i,k,l, kde k<l+c_i
pre vsetky i,k,l, kde k<l+c_i
-
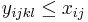 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
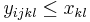 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
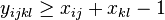 pre vsetky i,j,k,l, kde i<k, j<l
pre vsetky i,j,k,l, kde i<k, j<l
-
Na zamyslenie:
- Aka bude velkost programu ako funkcia n a m?
- Co ak nie vsetky jadra navzajom interaguju? Mozeme na velkosti programu usetrit?
- Preco asi vobec autori zaviedli jadra a ako by sme zmenili program, ak by sme chceli uvazovat kazdu aminokyselinu zvlast?
Zdroj:
- Jinbo Xu, Ming Li, Dongsup Kim, and Ying Xu. "RAPTOR: optimal protein threading by linear programming." Journal of bioinformatics and computational biology 1, no. 01 (2003): 95-117. [32]
Zhlukovanie (clustering)
- Máme vstupné dáta, väčšinou ako vektory dĺžky n
- Snažíme sa ich rozdeliť do skupín tak, aby dáta v rámci skupiny boli podobné a medzi skupinami rôzne
Využitie v bioinformatike:
- hľadanie génov s podobným profilom expresie
- hľadanie skupín pacientov s podobným profilom expresie génov (objavovanie podtypov nejakej choroby)
- hľadanie rodín podobných proteínov
- automatická segmentácia obrázkov (napríklad rozlíšiť jednotlivé políčka microarray alebo gelu od pozadia)
Na prednáške sme videli hierarchické zhlukovanie, ktoré z dát vytvorilo strom. Dnes si ukážeme zhlukovanie, ktoré sa snaží dáta rozdeliť na k skupín, kde k je vopred daný parameter.
K-Means
- pozri tiež prezentáciu pdf
- Vstup: Body
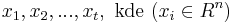 a počet zhlukov k
a počet zhlukov k
- Výstup: Rozdelenie do k zhlukov takéto:
- - priradenie bodu k zhluku
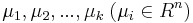 - centrá každého zhluku centroidy
- centrá každého zhluku centroidy
- Úloha: minimalizovať chybovú funkciu
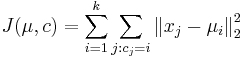 (inými slovami, počítam ako ďaleko (Euklidovská vzdialenosť) je každý bod od svojho centroidu?)
(inými slovami, počítam ako ďaleko (Euklidovská vzdialenosť) je každý bod od svojho centroidu?)
Algoritmus
Heuristika, ktorá nenájde vždy najlepšie zhlukovanie. Začne z nejakého zhlukovania a postupne ho zlepšuje. Pozri aj clanok na Wikipedii
- inicializácia: náhodne vyber k centroidov
- opakuj až do konvergencie:
- priraď každý bod najbližšiemu centroidu:
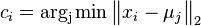
- vypočítaj nové centroidy: (spriemerujem všetky body v jednom zhluku)
- priraď každý bod najbližšiemu centroidu:
Úvod do bezkontextových gramatík
- Na modelovanie struktury RNA sa pouzivaju stochasticke bezkontextove gramatiky (bude na dalsej prednaske)
- Tie sú založené na bezkontextových gramatikách, ktoré mnohí poznáte z bakalárskeho štúdia
- Ak ste ich ale na bakalárskom štúdiu nebrali, pozrite si úvod na cvičeniach pre biológov
CB10
Nussinovovej algoritmus
Z cvičných príkladov na skúšku
- Vyplňte maticu dynamického programovania (Nussinovovej algoritmus) pre nájdenie najväčšieho počtu dobre uzátvorkovaných spárovaných báz v RNA sekvencii GAACUUCACUGA (dovoľujeme len komplementárne páry A-U, C-G) a nakreslite sekundárnu štruktúru, ktorú algoritmus našiel.
G A A C U U C A C U G A
0 0 0 1 1 2 3 3 3 4 4 4 G
0 0 0 1 2 2 2 2 3 4 4 A
0 0 1 1 1 2 2 2 3 4 A
0 0 0 0 1 1 1 2 3 C
0 0 0 1 1 1 2 3 U
0 0 1 1 1 2 3 U
0 0 0 1 2 2 C
0 0 1 1 1 A
0 0 1 1 C
0 0 1 U
0 0 G
0 A
- Ako by sme algoritmus upravili, aby dlzka slucky na konci helixu bola vzdy aspon 3?
Proteíny (dokončenie)
- Pozreli sme si na enzým Bis(5'-adenosyl)-triphosphatase na stránke http://www.uniprot.org/ pod názvom FHIT_HUMAN
- Tento enzým je vzdialene podobný na enzým galactose-1-phosphate uridylyltransferase (GALT/GAL7)
- Jeho domény patria v databáze Pfam do toho istého klanu, ako doména proteínu FHIT
- Skúsme nájsť túto podobnosť v BLASTe: http://blast.ncbi.nlm.nih.gov/ v časti proteíny, zvoľme databázu Swissport, ako Query zadajme Accesion nášho proteínu P49789, spustime program PSI-BLAST
- V prvom kole PSI-BLAST spúšťa bežný BLASTP
- GAL gén (konkrétne GAL7_HAEIN, accession P31764) sa nachádza medzi výsledkami, ale má príliš vysokú E-value
- Spustíme teraz druhú iteráciu PSI-BLAST, ktorá zostaví profil z proteínov s nízkou E-value v prvej iterácii
- Aká je E-value nájdeného zarovnania?
Sekvenčné motívy, program MEME
- Vazobne miesta transkripcnych faktorov sa casto reprezentuju ako sekvencne motivy
- Ak mame skupinu sekvencii, mozeme hladat motiv, ktory maju spolocny
- Znamy program na tento problem je MEME
- Chodte na stranku http://meme-suite.org/
- Zvolte nastroj MEME a v casti Input the primary sequences zvolte Type in sequences a zadajte tieto sekvencie (treba najskor ulozit do suboru)
- Pozrite si ostatne nastavenia. Co asi robia?
- Ak server pocita dlho, mozete si pozriet vysledky tu
Kvasinkové transkripčné faktory v SGD
- Yeast genome database SGD obsahuje pomerne podrobne stranky pre jednotlive transkripcne faktory
- Pozrime si stranku pre transkripcny faktor GAL4 [33]
RNA štruktúra
- Znama databaza rodin RNA genov je Rfam: http://rfam.xfam.org/
- Najdite si v nej rodinu RF00015 (U4 spliceosomal RNA)
- V casti Secondary structure si mozete pozriet obrazky farebne kodovane podla roznych kriterii
- Skuste pochopit, co jednotlive obrazky a ich farby znamenaju
- Jedna z mnohych ludskych kopii je tato:
AGCTTTGCGCAGTGGCAGTATCGTAGCCAATGAGGTTTATCCGAGGCGCG ATTATTGCTAATTGAAAACTTTTCCCAATACCCCGCCATGACGACTTGAA ATATAGTCGGCATTGGCAATTTTTGACAGTCTCTACGGAGA
- Skuste ju najst v ludskom genome nastrojom BLAT v UCSC genome browseri
- Pozrite si tracky GENCODE genes, conservation, RepeatMasker v jej okoli
- Vo verzii hg19 (kam sa viete z inej verzii dostat cez horne menu View->In Other Genomes) je track "CSHL Sm RNA-seq" ktory obsahuje RNASeq kratkych RNA z roznych casti buniek, zapnite si v jeho nastaveniach aj zobrazenie RNA z jadra (nucleus)
- Zadajte sekvenciu na RNAfold serveri [34]
- Ak vypocet dlho trva, pozrite si vysledok tu
- Podoba sa na strukturu zobrazenu v Rfame? v com sa lisi?
- RNA dizajn: mozete sa skusit zahrat na stranke http://www.eternagame.org/web/
CI11
RNA struktura
- Opakovanie Nussinovovej algoritmu
Z cvičných príkladov na skúšku
- Vyplnte maticu dynamického programovania (Nussinovovej algoritmus) pre nájdenie najväčšieho počtu dobre uzátvorkovaných spárovaných báz v RNA sekvencii GAACUUCACUGA (dovoľujeme len komplementárne páry A-U, C-G) a nakreslite sekundárnu štruktúru, ktorú algoritmus našiel.
G A A C U U C A C U G A
0 0 0 1 1 2 3 3 3 4 4 4 G
0 0 0 1 2 2 2 2 3 4 4 A
0 0 1 1 1 2 2 2 3 4 A
0 0 0 0 1 1 1 2 3 C
0 0 0 1 1 1 2 3 U
0 0 1 1 1 2 3 U
0 0 0 1 2 2 C
0 0 1 1 1 A
0 0 1 1 C
0 0 1 U
0 0 G
0 A
Rozsirenia Nussinovovej algoritmu
- lahke: kazdy par i,j musi mat vzdialenost |i-j|>=3 (RNA sa na kratsom useku nevie ohnut o 180 stupnov)
- tazsie (bolo s hintom na skuske): chceme davat skore iba "stackovanym parom", t.j. ak i a j aj i+1 a j-1 su sparovane, dostaneme +1, osamotene pary nedostavaju ziadne skore. Úlohou je opäť pre danú sekvenciu nájsť dobre uzátvorkovanú štruktúru s maximálnym skóre.
- pomocka: pouzijeme dve tabulky A a B, pričom A[i,j] obsahuje maximálne skóre pre podreťazec X[i...j] a B[i...j] obsahuje maximálne skóre pre podreťazec X[i...j], za predpokladu, že X[i] a X[j] sú spárované v štruktúre (táto hodnota je definovaná iba pre i a j, kde sú X[i] a X[j] komplementárne).
Stochasticke bezkontextove gramatiky
- Ako asi funguje algoritmus, ktory hlada najpravdepodobnejsie odvodenie?
- Je najpravdepodobnejsie odvodenie to iste ako najpravdepodobnejsia sekundarna struktura pri gramatike z prednasky?
- Viete zmenit gramatiku tak aby to bolo to iste?
Úvod do bioinformatických databáz a on-line nástrojov
NCBI, Genbank, Pubmed, blast
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/
- Zhromazduje verejne pristupne data z molekularnej biologie
- Mozeme hladat klucove slova v roznych databazach
- Pubmed: databaza clankov, napr. najdime phastcons
- Gene: najdime DNA polymerazu
- BLAST: najdime nasledujucu sekvenciu v genome kurata (zvoľme nucleotide blast, database others a z menu reference genomic sequence, organism chicken (taxid:9031), program blastn)
- Ide o osekvenovany kusok ludskej mRNA, kde v kuracom genome sme nasli homolog, ake ma dlzku, skore, E-value, % zhodnych baz?
AACCATGGGTATATACGACTCACTATAGGGGGATATCAGCTGGGATGGCAAATAATGATTTTATTTTGAC TGATAGTGACCTGTTCGTTGCAACAAATTGATAAGCAATGCTTTCTTATAATGCCAACTTTGTACAAGAA AGTTGGGCAGGTGTGTTTTTTGTCCTTCAGGTAGCCGAAGAGCATCTCCAGGCCCCCCTCCACCAGCTCC GGCAGAGGCTTGGATAAAGGGTTGTGGGAAATGTGGAGCCCTTTGTCCATGGGATTCCAGGCGATCCTCA CCAGTCTACACAGCAGGTGGAGTTCGCTCGGGAGGGTCTGGATGTCATTGTTGTTGAGGTTCAGCAGCTC CAGGCTGGTGACCAGGCAAAGCGACCTCGGGAAGGAGTGGATGTTGTTGCCCTCTGCGATGAAGATCTGC AGGCTGGCCAGGTGCTGGATGCTCTCAGCGATGTTTTCCAGGCGATTCGAGCCCACGTGCAAGAAAATCA GTTCCTTCAGGGAGAACACACACATGGGGATGTGCGCGAAGAAGTTGTTGCTGAGGTTTAGCTTCCTCAG TCTAGAGAGGTCGGCGAAGCATGCAGGGAGCTGGGACAGGCAGTTGTGCGACAAGCTCAGGACCTCCAGC TTTCGGCACAAGCTCAGCTCGGCCGGCACCTCTGTCAGGCAGTTCATGTTGACAAACAGGACCTTGAGGC ACTGTAGGAGGCTCACTTCTCTGGGCAGGCTCTTCAGGCGGTTCCCGCACAAGTTCAGGACCACGATCCG GGTCAGTTTCCCCACCTCGGGGAGGGAGAACCCCGGAGCTGGTTGTGAGACAAATTGAGTTTCTGGACCC CCGAAAAGCCCCCACAAAAAGCCG
Uniprot
- Prehladnejsi pohlad na proteiny, vela linkov na ine databazy, cast vytvarana rucne
- Pozrieme sa na enzým Bis(5'-adenosyl)-triphosphatase
- Nájdime ho na stránke http://www.uniprot.org/ pod názvom FHIT_HUMAN
- Pozrime si podrobne jeho stránku, ktoré časti boli predpovedané bioinformatickými metódami z prednášky?
- Všimnime si Pfam doménu a pozrime si jej stránku
UCSC genome browser
(nerobili sme, uvidíte na predmete Genomika)
- http://genome.ucsc.edu/
- On-line grafický nástroj na prezeranie genómov
- Konfigurovateľný, veľa možností, prijemne pouzivatelske rozhranie
- Moznost stiahnut data vhodne na dalsie spracovanie alebo zobrazit vlastne data
- Pomerne málo organizmov
- doraz hlavne na ludsky genom
Základy
- Adresa http://genome.ucsc.edu/
- Hore v modrom menu zvoľte Genomes, potom zvoľte ľudský genóm. Do okienka search term zadajte HOXA2. Vo výsledkoch hľadania (UCSC genes) zvoľte gén homeobox A2 na chromozóme 7.
- Pozrime si spolu túto stránku
- V hornej časti sú ovládacie prvky na pohyb vľavo, vpravo, približovanie, vzďaľovanie
- Pod tým schéma chromozómu, červeným vyznačená zobrazená oblasť
- Pod tým obrázok vybranej oblasti, rôzne tracky
- Pod tým zoznam všetkých trackov, dajú sa zapínať, vypínať a konfigurovať
- Po kliknutí na obrázok sa často zobrazí ďalšia informácia o danom géne alebo inom zdroji dát
- V génoch exony hrubé, UTR tenšie, intróny vodorovné čiary
- Po kliknutí na gén alebo inú časť nejakého tracku väčšinou o ňom dostaneme viac informácií. Kliknutim na listu ku tracku (lavy okraj obazku) sa dozviete viac o tracku a mozete nastavovat parametre zobrazenia
Sekvenovanie
- Hore v modrom menu zvoľte Genomes
- Na ďalšej stránke zvoľte človeka a v menu Assembly zistite, kedy boli pridané posledné dve verzie ľudského genómu (hg19 a hg38)
- Na tej istej stránke dole nájdete stručný popis zvolenej verzie genómu. Pre ktoré oblasti genómu máme v hg38 najviac alternatívnych verzií?
- Zadajte región chr21:31,250,000-31,300,000 v hg19 [35]
- Zapnite si tracky Mapability a RepeatMasker na "full"
- Mapability: nakoľko sa daný úsek opakuje v genóme a či teda vieme jednoznačne jeho ready namapovať pri použití Next generation sequencing
- Ako a prečo sa pri rôznych dĺžkach readov líšia? (Keď kliknete na linku "Mapability", môžete si prečítať bližšie detaily.)
- Približne v strede zobrazeného regiónu je pokles mapovateľnosti. Akému typu opakovania zodpovedá? (pozrite track RepeatMasker)
- Zapnite si tracky "Assembly" a "Gaps" a pozrite si región chr2:110,000,000-110,300,000 v hg19. [36] Aká dlhá je neosekvenovaná medzera (gap) v strede tohto regiónu? Približnú veľkosť môžete odčítať z obrázku, presnejší údaj zistíte kliknutím na čierny obdĺžnik zodpovedajúci tejto medzere (úplne presná dĺžka aj tak nebola známa, nakoľko nebola osekvenovaná).
- Cez menu položku View, In other genomes si pozrite, ako zobrazený úsek vyzerá vo verzii hg38. Ako sa zmenila dĺžka z pôvodných 300kb?
- Prejdite na genóm Rhesus, verzia rheMac2, región chr7:59,022,000-59,024,000 [37], zapnite si tracky Contigs, Gaps, Quality scores
- Aké typy problémov v kvalite sekvencie v tomto regióne vidíte?
Komparativna genomika
- V casti multiz alignments vidite zarovnania k roznym inym genomom (da sa zapinat, ze ku ktorym). Mozete si pozriet, ako sa uroven zarovnania zmeni ked sa priblizujeme a vzdalujeme (zoom in/zoom out).
- Ked sa priblizite na uroven "base", t.j. zobrazenych cca 100bp, v obdlzniku multiz alignment uvidite zarovnanie s homologickym usekom v inych genomoch.
- V casti conservation by PhyloP vidime graf toho, ako silne su zachovane jednotlive stlpce zarovnania
- Da sa zapnut track Placental Chain/Net a pozriet sa na ktorych chromozomoch je ortologicky usek v inych genomoch
Blat
- Choďte na UCSC genome browser (http//genome.ucsc.edu/), na modrej lište zvoľte BLAT, zadajte DNA sekvenciu vyssie a hľadajte ju v ľudskom genóme. Akú podobnosť (IDENTITY) má najsilnejší nájdený výskyt? Aký dlhý úsek genómu zasahuje? (SPAN). Všimnite si, že ostatné výskyty sú oveľa kratšie.
- V stĺpci ACTIONS si pomocou Details môžete pozrieť detaily zarovnania a pomocou Browser si pozrieť príslušný úsek genómu.
- V tomto úseku genómu si zapnite track Vertebrate net na full a kliknutím na farebnú čiaru na obrázku pre tento track zistite, na ktorom chromozóme kuraťa sa vyskytuje homologický úsek.
- Skusme tu istu sekvenciu namapovat do genomu sliepky: stlacte najprv na hornej modrej liste Genomes, zvolte Vertebrates a Chicken a potom na hornej liste BLAT. Do okienka zadajte tu istu sekvenciu. Akú podobnosť a dĺžku má najsilnejší nájdený výskyt teraz? Na ktorom je chromozóme?
- Ako sa to porovna s hodnotami, ktore sme dostali pomocou BLASTu na NCBI?
Práca s tabuľkami, sťahovanie anotácií
- Položka Tables na hornej lište umožnuje robiť rafinované veci s tabuľkami, ktoré obsahujú súradnice génov a pod.
- Základná vec: vyexportovať napr. všetky gény v zobrazenom výseku v niektorom formáte:
- sequence: fasta súbor proteínov, génov alebo mRNA s rôznymi nastaveniami
- GTF: súradnice
- Hyperlinks to genome browser: klikacia stránka
- Namiesto exportu si môžeme pozrieť rôzne štatistiky
- Zložitejšie: prienik dvoch tabuliek, napr. gény, ktoré sú viac než 50% pokryté simple repeats
- V intersection zvolíme group: Variation and repeats, track: RepeatMasker, nastavíme records that have at least 50% overlap with RepeatMasker
- V summary/statistics zistíme, kolko ich je v genóme, môžeme si ich preklikať cez Hyperlinks to genome browser
- Filter na tabuľku, napr. gény, ktoré majú v názve ribosomal (postup pre drozofilu):
- V casti hg19.kgXref based filters políčko description dáme *ribosomal*
CB11
Zhrnutie semestra
- vid prezentacia k cviceniu
Uvod do teorie grafov
- vid prezentacia k cviceniu
CI12
Gibbs sampling, Gibbsovo vzorkovanie vseobecne
- Cielove rozdelenie ma n premennych
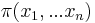
- V kazdom kroku vzorkujeme jednu premennu z podmienenej pravdepodobnosti
- Ostatne hodnoty nechame rovnake ako v predchadzajucom kroku
- Premennu zvolime nahodne alebo periodicky
- Vzorky nie su nezavisle, no vieme dokazat nieco o konvergencii k (pozri nizsie)
Markov chain Monte Carlo MCMC
- Chceme generovať náhodné vzorky z nejakeho cieloveho rozdelenia , ale toto rozdelenie prilis zlozite.
- Zostavime ergodicky Markovov retazec, ktoreho stacionarne rozdelenie je rozdelenie , tak aby sme efektivne vedeli vzorkovat
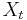 ak vieme
ak vieme 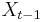 .
.
- Ak zacneme z lubovolneho bodu, po urcitom case t rozdelenie priblizne
- Ale za sebou iduce vzorky nie su nezavisle!
- Vieme vsak odhadovat ocakavane hodnoty roznych velicin
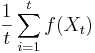 konverguje k
konverguje k
Určovanie štruktúry populácie
- Pritchard JK, Stephens M, Donnelly P (June 2000). "Inference of population structure using multilocus genotype data". Genetics 155 (2): 945–59. PMID 10835412.
- Majme N haploidnych jedincov (lahko sa rozsiri aj na genotypy pri diploidnych jedincoch), L genotypovanych SNPov, SNPy navzajom nezavisle (v stave LE), pocet populacii K
- X[i,l] - haplototyp jedinca i v SNPe l (zvacsa binarna premenna)
- Z[i,l] - z ktorej subpopulacie pochadza alela X[i,l] (cislo z {1...k})
- Q[i,k] - aka cast genomu jedinca i pochadza z populacie k (realne cislo)
- P[k,l,a] - frekvencia alely a v SNPe l v populacii k (realne cislo)
- P pochadza z nejakeho apriorneho rozdelenia, napr. rovnomerne rozdelenie, Dirichletovo rozdelenie, nezavisle pre kazde k,l
- Podobne Q (nezavisle pre kazde i)
- Pr(Z[i,l] = k|P,Q) = Q[i,k] a Pr(Z|P,Q) je sucin takychto clenov
- Pr(X[i,l] = a|Z,P,Q) = P[Z[i,l],l,a] a Pr(X|Z,P,Q) je sucin takychto clenov (SNPy nezavisle)
- To nam urcuje Pr(P,Q,Z,X) = Pr(P)Pr(Q)Pr(Z|P,Q)Pr(X|Z,P,Q)
- My chceme Pr(Q|X)
Algoritmus Gibbsovho vzorkovania:
- Zvol pociatocne
- Opakuj:
- Zvol nahodne
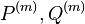 z
z 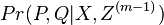
- Zvol nahodne
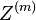 z
z 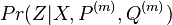
- Zvol nahodne
- Vzorce v clanku
- Mierna komplikacia: ak aproximujeme
![E[Q[i,k]|X]](/vyuka/mbi/images/math/c/7/b/c7b0785e7f607e83ca3a22f781a1a8e5.png) pomocou priemeru
pomocou priemeru 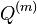 , mali by sme dostat 1/K kvoli symetrii (K! symetrickych rieseni)
, mali by sme dostat 1/K kvoli symetrii (K! symetrickych rieseni)
- Nastastie sa Gibbsovo vzorkovanie malokedy presuva medzi roznymi oznackovaniami tych istych populacii
- Inak musime pouzit nejako preznacit populacie vo vysledku aby boli ekvivalentne v roznych vzorkach