CB04: Rozdiel medzi revíziami
(→Uniprot pre projekty) |
|||
| (18 intermediate revisions by the same user not shown) | |||
| Riadok 1: | Riadok 1: | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
==Skórovacie matice== | ==Skórovacie matice== | ||
| Riadok 46: | Riadok 28: | ||
===Cvičenie pri počítači=== | ===Cvičenie pri počítači=== | ||
| − | * Stiahnite si súbor [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb05/scoring.ods] | + | * Stiahnite si súbor nižšie, uložte si ho a otvorte v Exceli / OpenOffice / LibreOffice |
| + | ** [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb05/scoring.ods ODS formát] | ||
| + | ** [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb05/scoring.xlsx XLSX formát for Excel] | ||
| + | ** [http://compbio.fmph.uniba.sk/vyuka/mbi-data/cb05/scoring-en.xlsx XLSX English version] | ||
* V záložke Matica vyplňte do žltej oblasti vzorce na výpočet pravdepodobnosti krátkeho zarovnania, logaritmus pomeru pravdepodobnosti a súčet skóre, pričom vo vzorcoch použijete odkazy na políčka v riadkoch 9-13, stĺpcoch B a E | * V záložke Matica vyplňte do žltej oblasti vzorce na výpočet pravdepodobnosti krátkeho zarovnania, logaritmus pomeru pravdepodobnosti a súčet skóre, pričom vo vzorcoch použijete odkazy na políčka v riadkoch 9-13, stĺpcoch B a E | ||
| − | * Súčet skóre by mal zhruba rovný desaťnásobku logaritmu pomeru - prečo vidíme rozdiely? | + | * Súčet skóre by mal byť zhruba rovný desaťnásobku logaritmu pomeru - prečo vidíme rozdiely? |
* Potom skúšajte meniť %GC a %identity v horných riadkoch tabuľky a pozrite sa, ako to ovplyvní skórovanie. Výsledné skóre zo stĺpca E ručne prepíšte (bez formúl) do tabuľky v záložke Výsledky. Prečo nastávajú také zmeny ako vidíte? | * Potom skúšajte meniť %GC a %identity v horných riadkoch tabuľky a pozrite sa, ako to ovplyvní skórovanie. Výsledné skóre zo stĺpca E ručne prepíšte (bez formúl) do tabuľky v záložke Výsledky. Prečo nastávajú také zmeny ako vidíte? | ||
| − | |||
==Praktické cvičenie pri počítači: dotploty== | ==Praktické cvičenie pri počítači: dotploty== | ||
| Riadok 76: | Riadok 60: | ||
** Pozrite si vysledok ako Dotplot, '''kolko opakovani Alu ste nasli? Preco je jedno cervene? ''' | ** Pozrite si vysledok ako Dotplot, '''kolko opakovani Alu ste nasli? Preco je jedno cervene? ''' | ||
** Pozrite si Raw: blast, '''na kolko percent sa podoba najpodobnejsia a na kolko druha najpodobnejsia kopia?''' | ** Pozrite si Raw: blast, '''na kolko percent sa podoba najpodobnejsia a na kolko druha najpodobnejsia kopia?''' | ||
| + | |||
| + | ==Dotplot celých kvasinových genómov== | ||
| + | * Na stránke https://dgenies.toulouse.inra.fr/run (based on minimap2 program) | ||
| + | * Zadáme URL dvoch genómov z NCBI: | ||
| + | ** Candida albicans https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/182/965/GCF_000182965.3_ASM18296v3/GCF_000182965.3_ASM18296v3_genomic.fna.gz | ||
| + | ** Candida dubliniensis https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/026/945/GCF_000026945.1_ASM2694v1/GCF_000026945.1_ASM2694v1_genomic.fna.gz | ||
| + | * Predpočítaný výsledok https://dgenies.toulouse.inra.fr/result/CL9nR_20231012150020 | ||
| + | * Iná dvojica: | ||
| + | ** Magnusiomyces ingens ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/uid/UIDE01.fasta.gz [https://www.ebi.ac.uk/ena/browser/view/GCA_900497715?show=blobtoolkit] | ||
| + | ** Saprochaete ingens ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/cab/CABVLU01.fasta.gz [https://www.ebi.ac.uk/ena/browser/view/GCA_902498895] | ||
==Príklady praktických programov== | ==Príklady praktických programov== | ||
| Riadok 85: | Riadok 79: | ||
* Balicek emboss, obsahuje programy na klasicke dynamicke programovanie (needle - globalne, water - lokalne), najdu sa na stranke EBI http://www.ebi.ac.uk/Tools/psa/ | * Balicek emboss, obsahuje programy na klasicke dynamicke programovanie (needle - globalne, water - lokalne), najdu sa na stranke EBI http://www.ebi.ac.uk/Tools/psa/ | ||
* porovnanie lokalneho a globalneho zarovnania | * porovnanie lokalneho a globalneho zarovnania | ||
| − | ** Dva proteiny | + | ** Dva proteiny z rôznych kvasiniek zarovnáme lokálne, globálne a globálne s tým, že neplatíme za medzery na koncoch |
| − | + | * sekvencie a vysledne zarovnania: [[CB-aln-dp]] | |
| − | + | * vo vysledku si vsimnime, kolko ma kazde z nich %identity, %gaps, a kam sa zarovna sekvencia IRESPLGG ktora je na pozicii 29 v prvom a 30 v druhom proteine | |
| − | + | ||
| − | + | ||
| − | * sekvencie a vysledne zarovnania: [[ | + | |
| − | * vo vysledku si vsimnime, kolko ma kazde z nich %identity, %gaps, a kam sa zarovna sekvencia na pozicii | + | |
<pre> | <pre> | ||
Lokalne zarovnanie | Lokalne zarovnanie | ||
| − | Length: | + | Length: 588 |
| − | Identity: | + | Identity: 170/588 (28.9%) |
| − | Similarity: | + | Similarity: 270/588 (45.9%) |
| − | Gaps: | + | Gaps: 116/588 (19.7%) |
| − | Score: | + | Score: 611.0 |
| − | + | MCA_00027_1 29-568 (z 595) | |
| − | + | RKM3_YEAST 30-549 (z 552) | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | 549 | + | |
| − | + | ||
| − | + | ||
Globalne zarovnanie | Globalne zarovnanie | ||
| − | Length: | + | Length: 650 |
| − | Identity: | + | Identity: 178/650 (27.4%) |
| − | Similarity: | + | Similarity: 282/650 (43.4%) |
| − | Gaps: | + | Gaps: 153/650 (23.5%) |
| − | Score: | + | Score: 588.5 |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
Globalne zarovnanie s nulovou penaltou za medzeru na koncoch | Globalne zarovnanie s nulovou penaltou za medzeru na koncoch | ||
| − | Length: | + | Length: 651 |
| − | Identity: | + | Identity: 177/651 (27.2%) |
| − | Similarity: | + | Similarity: 282/651 (43.3%) |
| − | Gaps: | + | Gaps: 155/651 (23.8%) |
| − | Score: | + | Score: 608.0 |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</pre> | </pre> | ||
| Riadok 213: | Riadok 173: | ||
Sbjct 241 ILLISFLIFLIVG 253 | Sbjct 241 ILLISFLIFLIVG 253 | ||
</pre> | </pre> | ||
| + | |||
| + | ==Uniprot pre projekty== | ||
| + | * Prehladny pohlad na proteiny, vela linkov na ine databazy, cast vytvarana rucne | ||
| + | <!-- * Pozrime si ľudský proteín PTPRZ1 v databáze Uniprot http://www.uniprot.org/ --> | ||
| + | * Pozrieme sa na známy koronavírusový proteín Spike | ||
| + | ** Nájdime ho na stránke http://www.uniprot.org/ pod názvom SPIKE_SARS2 | ||
| + | ** Pozrime si podrobne jeho stránku, ktoré časti boli predpovedané bioinformatickými metódami z prednášky? | ||
| + | ** Všimnime si niektorú Pfam doménu a pozrime si jej stránku | ||
| + | |||
| + | ==Praktické cvičenie pri počítači: BLAT vs BLAST== | ||
===BLAT, chains, nets v UCSC browseri=== | ===BLAT, chains, nets v UCSC browseri=== | ||
| Riadok 218: | Riadok 188: | ||
** Vhodne pouzitie: zarovnanie mRNA ku genomu, presne urcenie suradnic nejakej sekvencie, a pod. | ** Vhodne pouzitie: zarovnanie mRNA ku genomu, presne urcenie suradnic nejakej sekvencie, a pod. | ||
* Net tracky v UCSC genome browseri nam umoznuju prechadzat medzi homologickymi oblastami roznych genomov | * Net tracky v UCSC genome browseri nam umoznuju prechadzat medzi homologickymi oblastami roznych genomov | ||
| − | |||
| − | |||
===BLAT/BLAST=== | ===BLAT/BLAST=== | ||
Aktuálna revízia z 10:57, 16. november 2023
Obsah
Skórovacie matice
Chceme určiť skórovaciu schému pre zarovnávanie dvoch DNA sekvencií (bez medzier). Máme dva modely, každý z nich vie vygenerovať 2 zarovnané sekvencie dĺžky n.
Model R (random) reprezentuje nezávislé náhodne sekvencie
- Použijeme naše vrece s guličkami označenými A,C,G,T, pričom guličiek označených A je 30%, C 20%, G 20% a T 30%.
- Vytiahneme guličku, zapíšeme si písmeno, hodíme ju naspäť, zamiešame a opakujeme s ďalším písmenom atď až kým nevygenerujeme n písmen pre jednu sekvenciu a n písmen pre druhú
- Máme jednu sekvenciu ACT a druhú ACC. Aká je pravdepodobnosť, že práve tieto sekvencie vygenerujeme v našom modeli R?
- Nezávislé udalosti pre jednotlivé písmená, t.j. Pr(X1=A)*Pr(X2=C)*Pr(X3=T)*Pr(Y1=A)*Pr(Y2=C)*Pr(Y3=C) = 0.3*0.2*0.3*0.3*0.2*0.2 = 0.000216
- Spolu máme v modeli
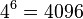 možností ako vygenerovať 2 DNA sekvencie dĺžky 3
možností ako vygenerovať 2 DNA sekvencie dĺžky 3
Model H (homolog) reprezentuje zarovnanie vzájomne súvisiacich sekvencií
- máme vrece, v ktorom je napr.
- po 21% guličiek označených AA, TT
- po 14% označených CC, GG
- po 2.4% označených AC, AG, CA, CT, GA, GT, TC, TG
- po 3.6% označených AT, TA
- po 1.6% označených CG, GC.
- Spolu máme 70% guličiek označených rovnakými písmenami, 30% rôznymi
- n krát z vreca vytiahneme guličku a písmená píšeme ako stĺpce zarovnania A1, A2,.., An.
- aká je pravdepodobnosť, ze dostaneme ACT zarovnané s ACC?
- Pr(A1=AA)*Pr(A2=CC)*Pr(A3=TC) = 0.21*0.14*0.024 = 0.0007056
Skóre zarovnania je log Pr(zarovnania v H)/Pr(zarovnania v R), t.j. log (0.0007056 / 0.000216) = 0.514105 (pre desiatkový logaritmus)
- kladné skóre znamená, že model H lepšie zodpovedá dátam (zarovnaniu) ako model R
- záporné skóre znamená, že model R lepšie zodpovedá dátam
Cvičenie pri počítači
- Stiahnite si súbor nižšie, uložte si ho a otvorte v Exceli / OpenOffice / LibreOffice
- V záložke Matica vyplňte do žltej oblasti vzorce na výpočet pravdepodobnosti krátkeho zarovnania, logaritmus pomeru pravdepodobnosti a súčet skóre, pričom vo vzorcoch použijete odkazy na políčka v riadkoch 9-13, stĺpcoch B a E
- Súčet skóre by mal byť zhruba rovný desaťnásobku logaritmu pomeru - prečo vidíme rozdiely?
- Potom skúšajte meniť %GC a %identity v horných riadkoch tabuľky a pozrite sa, ako to ovplyvní skórovanie. Výsledné skóre zo stĺpca E ručne prepíšte (bez formúl) do tabuľky v záložke Výsledky. Prečo nastávajú také zmeny ako vidíte?
Praktické cvičenie pri počítači: dotploty
Yass a dotploty
- Program Yass hlada lokalne zarovnania v DNA sekvenciach, zobrazuje vo forme dot plotov
- V novom okne/tabe si otvorte YASS server na adrese http://bioinfo.lifl.fr/yass/yass.php
- V dalsom okne si na stranke UCSC genome browseru si zobrazte oblast chr21:9,180,027-9,180,345 vo verzii hg38 ludskeho genomu [1]
- tento región obsahuje Alu repeat. Tieto opakovania tvoria cca 10% ľudského genómu, viac ako milión kópií
- zobrazte si DNA sekvenciu tohto useku takto: na hornej modrej liste zvolte View, potom v podmenu DNA, na dalsej obrazovke tlacidlo get DNA
- DNA sekvenciu Alu opakovania chceme zarovnat samu k sebe programom YASS
- DNA sekvenciu Alu opakovania skopirujte do okienka "Paste your sequences" v stranke Yass-u a dvakrat stlacte tlacidlo Select vedla okienka
- Nizsie v casti "Selected DNA sequence(s)" by sa Vam malo v oboch riadkoch objavit "Pasted file 1"
- Nizsie v casti "Parameters" zvolte "E-value threshold" 0.01 a stlacte "Run YASS"
- Vo vysledkoch si pozrite Dotplot, co z neho viete usudit o podobnosti jednotlivych casti Alu opakovania?
- Vo vysledkoch si pozrite Raw: blast, ake su suradnice opakujucej sa casti a kolko zarovnanie obsahuje zhod/nezhod/medzier? (Pozor, prve zarovnanie je cela sekvencia sama k sebe, druhe je asi to, co chcete)
- V genome browseri sa presunte na poziciu chr21:8,552,000-8,562,000 (10kb sekvencie na chromozome 21, s niekolkymi vyskytmi Alu) [2]
- Chceme teraz porovnat tento usek genomu so sekvenciou Alu pomocou YASSu
- Ako predtym si stiahnite DNA sekvenciu tohto useku
- V YASSe chodte sipkou spat na formular
- Skopirujte DNA sekvenciu do YASSoveho formulara, do okienka vpravo (vyznacit si ju mozete klavesovou kombinaciou Ctrl-A alebo Select All v menu Edit),
- V casti formulara Selected DNA sequence(s) stlacte Remove pri hornom riadku
- Pri pravom okienku, kam ste nakopirovali sekvenciu, stlacte Select
- Zase stlacte Run YASS
- Pozrite si vysledok ako Dotplot, kolko opakovani Alu ste nasli? Preco je jedno cervene?
- Pozrite si Raw: blast, na kolko percent sa podoba najpodobnejsia a na kolko druha najpodobnejsia kopia?
Dotplot celých kvasinových genómov
- Na stránke https://dgenies.toulouse.inra.fr/run (based on minimap2 program)
- Zadáme URL dvoch genómov z NCBI:
- Predpočítaný výsledok https://dgenies.toulouse.inra.fr/result/CL9nR_20231012150020
- Iná dvojica:
- Magnusiomyces ingens ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/uid/UIDE01.fasta.gz [3]
- Saprochaete ingens ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/cab/CABVLU01.fasta.gz [4]
Príklady praktických programov
Pozrime sa na niekolko nastrojov, vsimnime si, ake poskytuju nastavenia a co vypisuju na vystupe, dajme to do suvisu s prednaskami
- viacnasobne zarovnania neskor
Plné dynamické programovanie
- Balicek emboss, obsahuje programy na klasicke dynamicke programovanie (needle - globalne, water - lokalne), najdu sa na stranke EBI http://www.ebi.ac.uk/Tools/psa/
- porovnanie lokalneho a globalneho zarovnania
- Dva proteiny z rôznych kvasiniek zarovnáme lokálne, globálne a globálne s tým, že neplatíme za medzery na koncoch
- sekvencie a vysledne zarovnania: CB-aln-dp
- vo vysledku si vsimnime, kolko ma kazde z nich %identity, %gaps, a kam sa zarovna sekvencia IRESPLGG ktora je na pozicii 29 v prvom a 30 v druhom proteine
Lokalne zarovnanie Length: 588 Identity: 170/588 (28.9%) Similarity: 270/588 (45.9%) Gaps: 116/588 (19.7%) Score: 611.0 MCA_00027_1 29-568 (z 595) RKM3_YEAST 30-549 (z 552) Globalne zarovnanie Length: 650 Identity: 178/650 (27.4%) Similarity: 282/650 (43.4%) Gaps: 153/650 (23.5%) Score: 588.5 Globalne zarovnanie s nulovou penaltou za medzeru na koncoch Length: 651 Identity: 177/651 (27.2%) Similarity: 282/651 (43.3%) Gaps: 155/651 (23.8%) Score: 608.0
NCBI Blast
- NCBI BLAST http://blast.ncbi.nlm.nih.gov/ vela roznych nastrojov (porovnavanie DNA vs proteiny, pripadne translacia DNA na protein v 6 ramcoch)
- Heuristicky algoritmus, moze niektore zarovnania vynechat
- rozne nastavenia, vystup E-value
Low complexity masking: nepouzivat pri hladani jadier zarovnania regiony v ktorych sa velakrat opakuje ta ista aminokyselina
- Priklad (z ucebnice Zvelebil and Baum):
>sp|P04156|PRIO_HUMAN Major prion protein OS=Homo sapiens GN=PRNP PE=1 SV=1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV ILLISFLIFLIVG
- Hladajme v databaze Reference sequence (Refseq), organizmus human
Bez maskovania vypise napr aj toto zarovnanie:
>ref|NP_065842.1| serine/threonine-protein kinase TAO1 isoform 1 [Homo sapiens]
Length=1001
Score = 45.1 bits (105), Expect = 1e-06, Method: Composition-based stats.
Identities = 26/61 (43%), Positives = 27/61 (44%), Gaps = 11/61 (18%)
Query 38 YPGQGSPGGNRYPPQGGGG--WGQPHGG---GWGQPHGGG---WGQPHGGGWGQPHGGGWG 90
YPG G + P GG G WG P GG WG P GG WG P G G P G G
Sbjct 904 YPGAS---GWSHNPTGGPGPHWGHPMGGPPQAWGHPMQGGPQPWGHPSGPMQGVPRGSSMG 961
Score = 40.0 bits (92), Expect = 4e-05, Method: Composition-based stats.
Identities = 25/62 (40%), Positives = 25/62 (40%), Gaps = 10/62 (16%)
Query 26 PKPGGW--NTGGSRYPGQGSPGGNRYPPQGGGGWGQPHGGG---WGQPHGGGWGQPHGGGWG 82
P GW N G P G P G PPQ WG P GG WG P G G P G
Sbjct 905 PGASGWSHNPTGGPGPHWGHPMGG--PPQA---WGHPMQGGPQPWGHPSGPMQGVPRGSSMG 961
Ak zapneme maskovanie, toto zarovnanie uz nenajde, v zarovnani sameho so sebou sa objavia male pismena alebo Xka:
>ref|NP_000302.1|major prion protein preproprotein [Homo sapiens]
Length=253
Score = 520 bits (1340), Expect = 0.0, Method: Compositional matrix adjust.
Identities = 253/253 (100%), Positives = 253/253 (100%), Gaps = 0/253 (0%)
Query 1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYppqggggwgqp 60
MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP
Sbjct 1 MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQP 60
Query 61 hgggwgqphgggwgqphgggwgqphgggwgqgggTHSQWNKPSKPKTNMKHMagaaaaga 120
HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA
Sbjct 61 HGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGA 120
Query 121 vvgglggymlgsamsRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV 180
VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV
Sbjct 121 VVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCV 180
Query 181 NITIKQHtvttttkgenftetDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSsppv 240
NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV
Sbjct 181 NITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV 240
Query 241 illisfliflivG 253
ILLISFLIFLIVG
Sbjct 241 ILLISFLIFLIVG 253
Uniprot pre projekty
- Prehladny pohlad na proteiny, vela linkov na ine databazy, cast vytvarana rucne
- Pozrieme sa na známy koronavírusový proteín Spike
- Nájdime ho na stránke http://www.uniprot.org/ pod názvom SPIKE_SARS2
- Pozrime si podrobne jeho stránku, ktoré časti boli predpovedané bioinformatickými metódami z prednášky?
- Všimnime si niektorú Pfam doménu a pozrime si jej stránku
Praktické cvičenie pri počítači: BLAT vs BLAST
BLAT, chains, nets v UCSC browseri
- Program BLAT v UCSC browseri http://genome-euro.ucsc.edu/ rychlo vyhladava sekvencie v genome, ale nevie najst slabsie podobnosti
- Vhodne pouzitie: zarovnanie mRNA ku genomu, presne urcenie suradnic nejakej sekvencie, a pod.
- Net tracky v UCSC genome browseri nam umoznuju prechadzat medzi homologickymi oblastami roznych genomov
BLAT/BLAST
- Sekvencia uvedena nizsie vznikla pomocou RT-PCR na ľudských cDNA knižniciach
- Choďte na UCSC genome browser http://genome-euro.ucsc.edu/ , na modrej lište zvoľte BLAT, zadajte túto sekvenciu a hľadajte ju v ľudskom genóme. Akú podobnosť (IDENTITY) má najsilnejší nájdený výskyt? Aký dlhý úsek genómu zasahuje? (SPAN). Všimnite si, že ostatné výskyty sú oveľa kratšie.
- V stĺpci ACTIONS si pomocou Details môžete pozrieť detaily zarovnania a pomocou Browser si pozrieť príslušný úsek genómu.
- V tomto úseku genómu si zapnite track Vertebrate net na full a kliknutím na farebnú čiaru na obrázku pre tento track zistite, na ktorom chromozóme sliepky sa vyskytuje homologický úsek.
- Skusme tu istu sekvenciu zarovnat ku genomu sliepky programom Blat: stlacte najprv na hornej modrej liste Genomes, zvolte Vertebrates a Chicken a potom na hornej liste BLAT. Do okienka zadajte tu istu sekvenciu. Akú podobnosť a dĺžku má najsilnejší nájdený výskyt teraz? Na ktorom je chromozóme?
- Skúsme to isté v NCBI blaste: Choďte na http://blast.ncbi.nlm.nih.gov/ zvoľte nucleotide blast, database others a z menu reference genomic sequence, organism chicken (taxid:9031), program blastn
- Aká je dĺžka, identity a E-value najlepšieho zarovnania? Na ktorom je chromozóme?
RT PCR sekvencia z cvičenia vyššie
AACCATGGGTATATACGACTCACTATAGGGGGATATCAGCTGGGATGGCAAATAATGATTTTATTTTGAC TGATAGTGACCTGTTCGTTGCAACAAATTGATAAGCAATGCTTTCTTATAATGCCAACTTTGTACAAGAA AGTTGGGCAGGTGTGTTTTTTGTCCTTCAGGTAGCCGAAGAGCATCTCCAGGCCCCCCTCCACCAGCTCC GGCAGAGGCTTGGATAAAGGGTTGTGGGAAATGTGGAGCCCTTTGTCCATGGGATTCCAGGCGATCCTCA CCAGTCTACACAGCAGGTGGAGTTCGCTCGGGAGGGTCTGGATGTCATTGTTGTTGAGGTTCAGCAGCTC CAGGCTGGTGACCAGGCAAAGCGACCTCGGGAAGGAGTGGATGTTGTTGCCCTCTGCGATGAAGATCTGC AGGCTGGCCAGGTGCTGGATGCTCTCAGCGATGTTTTCCAGGCGATTCGAGCCCACGTGCAAGAAAATCA GTTCCTTCAGGGAGAACACACACATGGGGATGTGCGCGAAGAAGTTGTTGCTGAGGTTTAGCTTCCTCAG TCTAGAGAGGTCGGCGAAGCATGCAGGGAGCTGGGACAGGCAGTTGTGCGACAAGCTCAGGACCTCCAGC TTTCGGCACAAGCTCAGCTCGGCCGGCACCTCTGTCAGGCAGTTCATGTTGACAAACAGGACCTTGAGGC ACTGTAGGAGGCTCACTTCTCTGGGCAGGCTCTTCAGGCGGTTCCCGCACAAGTTCAGGACCACGATCCG GGTCAGTTTCCCCACCTCGGGGAGGGAGAACCCCGGAGCTGGTTGTGAGACAAATTGAGTTTCTGGACCC CCGAAAAGCCCCCACAAAAAGCCG